Mastering the SCF Iterative Process: A Comprehensive Guide from Theory to Troubleshooting for Computational Researchers
This guide provides a comprehensive overview of the Self-Consistent Field (SCF) iterative process, essential for Hartree-Fock and Kohn-Sham Density Functional Theory calculations.
Mastering the SCF Iterative Process: A Comprehensive Guide from Theory to Troubleshooting for Computational Researchers
Abstract
This guide provides a comprehensive overview of the Self-Consistent Field (SCF) iterative process, essential for Hartree-Fock and Kohn-Sham Density Functional Theory calculations. Covering foundational theory, practical methodologies, advanced troubleshooting techniques, and validation procedures, it addresses the complete workflow encountered by computational researchers. Special emphasis is placed on convergence acceleration strategies, stability analysis, and handling challenging systems relevant to drug development and biomedical research, enabling scientists to efficiently obtain reliable electronic structure solutions.
SCF Fundamentals: Understanding the Core Theory and Mathematical Framework
The Self-Consistent Field (SCF) method represents a cornerstone of modern computational chemistry and materials science, providing the fundamental algorithm for solving the quantum mechanical equations governing electronic structure. SCF theory encompasses both Hartree-Fock (HF) theory and Kohn-Sham Density Functional Theory (DFT), which together form the basis for most first-principles simulations of atoms, molecules, and materials [1] [2]. The origins of SCF theory date back to the late 1920s, shortly after the discovery of the Schrödinger equation in 1926 [3]. In 1927, Douglas Hartree introduced an approximate method to solve the Schrödinger equation by writing the wave function as a product of single-particle functions called orbitals, employing an iterative procedure he termed the "self-consistent field" method [4] [3].
The historical development of SCF theory represents a continuous refinement of these initial ideas. In 1930, Slater and Fock independently recognized that Hartree's method failed to satisfy the Pauli exclusion principle, leading to the introduction of an exchange term and the development of the Hartree-Fock method [4] [3]. The subsequent evolution continued with the Thomas-Fermi-Dirac model in 1930 [3], Slater's Xα method in 1951 [3], and the groundbreaking work of Hohenberg and Kohn in 1964, which established the theoretical foundation for modern density functional theory [3]. The introduction of the Kohn-Sham equations in 1965 provided a practical computational framework that remains the basis for most DFT calculations today [3]. This rich history demonstrates how SCF theory has continuously evolved over nearly a century, with each advancement building upon and refining the work of previous researchers.
Theoretical Foundations
The Schrödinger Equation and the Electron Correlation Problem
At the heart of quantum chemistry lies the many-electron Schrödinger equation, which describes the behavior of electrons in atoms and molecules. Theoretical physicist Paul Dirac famously noted in 1929 that "the underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble" [3]. This fundamental challenge arises primarily from the electron correlation problem—the complex, instantaneous interactions between electrons that make the Schrödinger equation computationally intractable for systems with more than a few electrons.
The SCF approach addresses this challenge through a mean-field approximation where each electron experiences the average field created by all other electrons, rather than their instantaneous interactions [4]. In both HF and KS-DFT, the ground-state wavefunction is expressed as a single Slater determinant of molecular orbitals (MOs) ψ: Φ₀ = 𝒜|ψ₁(1)ψ₂(2)…ψ_N(N)|, where 𝒜 is the antisymmetrizer [1]. The total electronic energy E = ⟨Ψ₀|Ĥ|Ψ₀⟩ is then minimized subject to orbital orthogonality constraints [1].
The Unified SCF Equation: Roothaan-Hall and Pople-Nesbet Formulations
The minimization of the total energy within a given basis set leads to a unified matrix equation that forms the core of SCF theory:
FC = SCE [1]
In this equation, C represents the matrix of molecular orbital coefficients, E is a diagonal matrix of the corresponding orbital eigenenergies, and S is the atomic orbital overlap matrix [1]. The Fock matrix F captures the effective one-electron energy operators and is defined as:
F = T + V + J + K [1]
Where:
- T is the kinetic energy matrix
- V is the external potential (typically nuclear-electron attraction)
- J is the Coulomb matrix representing electron-electron repulsion
- K is the exchange matrix arising from quantum mechanical exchange effects [1]
For restricted Hartree-Fock (RHF) formalism, the Fock matrix elements can be expressed as:
Fμν = Hμν^core + Jμν - Kμν [5]
Where the core Hamiltonian matrix elements Hμν^core = Tμν + Vμν consist of kinetic energy elements (Tμν) and nuclear attraction elements (Vμν) [5]. The Coulomb and exchange elements are given by:
Jμν = ∑λσ Pλσ (μν|λσ) Kμν = ½ ∑λσ Pλσ (μλ|νσ) [5]
Here, Pλσ represents the density matrix elements, and (μν|λσ) are the two-electron integrals [5].
For open-shell systems, the unrestricted form (UHF) introduces separate α and β density matrices:
Pμν^α = ∑a Cμa^α Cνa^α Pμν^β = ∑a Cμa^β Cνa^β [5]
The total electron density matrix is then P = P^α + P^β, leading to separate Fock matrices for α and β spins [5].
From Hartree-Fock to Kohn-Sham Density Functional Theory
While Hartree-Fock theory provides a rigorous foundation for electronic structure calculation, it suffers from a significant limitation: it neglects Coulomb correlation, treating electron interactions only in an average manner [4]. This limitation manifests in HF's inability to accurately capture London dispersion forces and other correlation-dependent phenomena [4].
Kohn-Sham Density Functional Theory addresses this limitation while maintaining a similar computational structure to Hartree-Fock. The key theoretical breakthrough came with the Hohenberg-Kohn theorems in 1964, which established that a method based on electron density alone could be exact [3]. Kohn and Sham subsequently introduced their famous equations in 1965, which provide a practical computational framework [3].
The Kohn-Sham model captures a large part of the DFT energy functional, with only one term—the exchange-correlation functional—remaining unknown [3]. The accuracy of Kohn-Sham DFT is therefore determined by the quality of the exchange-correlation functional approximation. The local density approximation (LDA) introduced by Kohn and Sham was the first such approximation, accurate for systems with slowly varying electron densities but limited for molecular systems [3]. The development of generalized gradient approximations (GGAs) in the 1980s represented a significant improvement, incorporating the density gradient to achieve useful chemical accuracy [3]. Hybrid functionals, introduced by Axel Becke in 1993, further improved accuracy by mixing a fraction of Hartree-Fock exchange with GGA functionals [3].
Table 1: Key Historical Developments in SCF Theory
| Year | Development | Key Researchers | Significance |
|---|---|---|---|
| 1926 | Schrödinger Equation | Erwin Schrödinger | Foundation of quantum mechanics |
| 1927 | Hartree Method | Douglas Hartree | First SCF iterative procedure |
| 1930 | Hartree-Fock Method | Slater and Fock | Incorporated Pauli exclusion principle |
| 1964 | Hohenberg-Kohn Theorems | Hohenberg and Kohn | Theoretical foundation for DFT |
| 1965 | Kohn-Sham Equations | Kohn and Sham | Practical computational framework for DFT |
| 1980s | Generalized Gradient Approximations (GGAs) | Becke, Perdew, others | First DFT approximations useful for chemistry |
| 1993 | Hybrid Functionals | Axel Becke | Mixed Hartree-Fock and DFT exchange |
| 1998 | Nobel Prize for DFT | Walter Kohn | Recognition of DFT's impact |
The SCF Iterative Process: Algorithms and Workflows
The SCF method is fundamentally an iterative procedure that progressively refines an initial guess of the electron density or molecular orbitals until self-consistency is achieved. The nonlinear nature of the Fock operator, which depends on its own solution through the density matrix, necessitates this iterative approach [4]. The quality of the initial guess significantly impacts the convergence behavior and success of the SCF procedure [1].
Initial Guess Strategies
Several sophisticated methods have been developed for generating initial guesses in SCF calculations:
- 'minao' guess: A superposition of atomic densities technique that projects the minimal basis onto the orbital basis set [1]
- 'atom' guess: Superposition of atomic densities employing spin-restricted atomic HF calculations [1]
- 'huckel' guess: A parameter-free Hückel guess based on atomic HF calculations [1]
- 'vsap' guess: Superposition of atomic potentials, available only for DFT calculations [1]
- '1e' guess: One-electron (core) guess that ignores interelectronic interactions [1]
- 'chk' guess: Reading orbitals from a checkpoint file of a previous calculation [1]
The accuracy of these initial guesses has been systematically assessed in recent literature, with the 'minao' guess serving as the default in many implementations like PySCF [1]. For challenging systems, a particularly effective strategy involves first computing the HF density matrix for a related system (such as an ionized species) and then using it as an initial guess for the target system [1].
Core SCF Algorithm
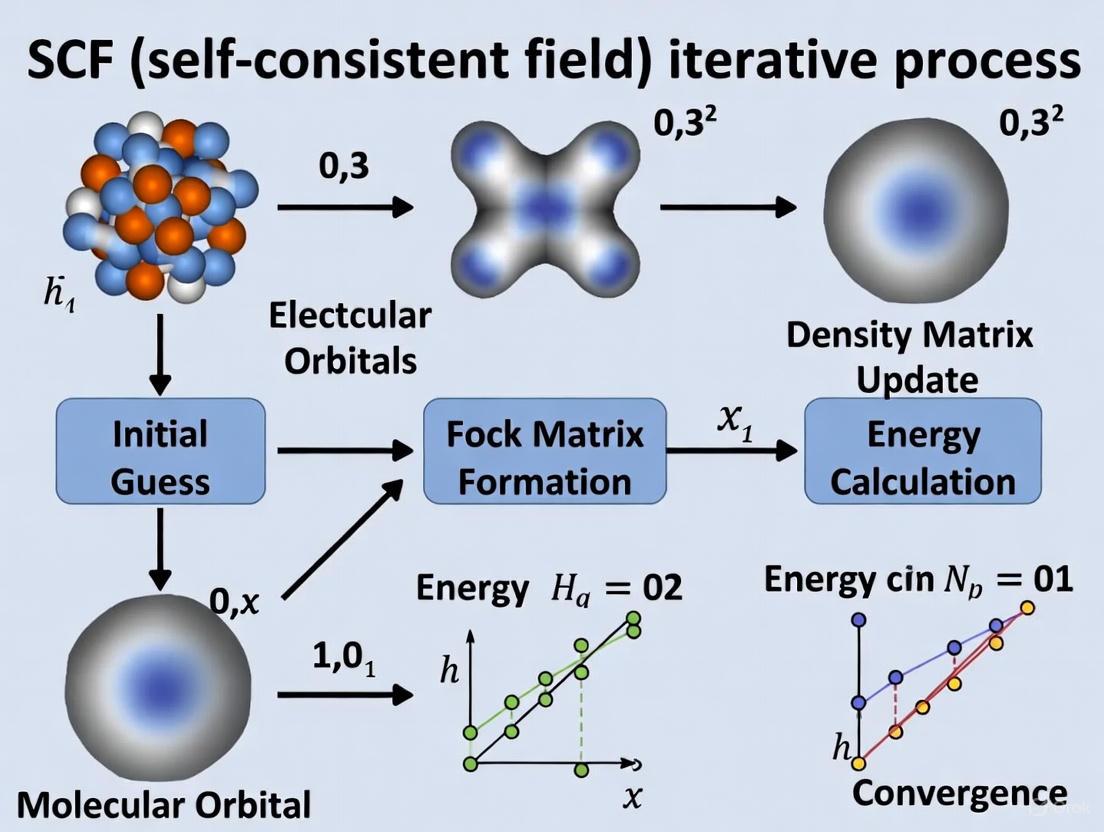
The following diagram illustrates the complete SCF iterative workflow, integrating the key components and decision points:
The SCF process begins with generating an initial guess for the density matrix or molecular orbitals, then proceeds through the core cycle of Fock matrix construction, solving the Roothaan-Hall equations, and forming a new density matrix. This cycle continues until convergence criteria are satisfied or the maximum number of iterations is reached. Following convergence, stability analysis is recommended to ensure the solution represents a true minimum rather than a saddle point [1].
Convergence Acceleration Algorithms
Due to the iterative nature of SCF calculations, convergence acceleration methods are essential for practical computations. Several algorithms have been developed for this purpose:
DIIS (Direct Inversion in the Iterative Subspace): The default method in many quantum chemistry codes, DIIS extrapolates the Fock matrix using information from previous iterations by minimizing the norm of the commutator [F, PS], where P is the density matrix and S is the overlap matrix [1] [6]. The DIIS coefficients are obtained by solving a constrained minimization problem [6]. Variants include EDIIS (Energy-DIIS) and ADIIS (Accelerated DIIS) [1] [6].
Geometric Direct Minimization (GDM): This approach takes steps in orbital rotation space that properly account for the hyperspherical geometry of that space, making it extremely robust for difficult-to-converge systems [6]. GDM is particularly recommended for restricted open-shell SCF calculations [6].
Second-Order SCF (SOSCF): Methods such as the co-iterative augmented hessian (CIAH) approach achieve quadratic convergence by utilizing orbital Hessian information [1]. These methods can be invoked when standard DIIS approaches fail.
Damping and Level Shifting: Simple yet effective techniques include damping (mixing a fraction of the previous Fock matrix with the new one) and level shifting (artificially increasing the energy gap between occupied and virtual orbitals) [1] [7].
Table 2: SCF Convergence Acceleration Methods
| Method | Algorithm Type | Key Features | Typical Applications |
|---|---|---|---|
| DIIS | Extrapolation | Minimizes error vector norm; Fast convergence | Default for most systems |
| GDM | Direct minimization | Robust; Accounts for orbital space geometry | Difficult cases; Restricted open-shell |
| SOSCF | Second-order | Uses orbital Hessian; Quadratic convergence | When DIIS fails; High accuracy |
| Damping | Stabilization | Mixes old and new Fock matrices | Initial cycles; Oscillatory cases |
| Level Shifting | Stabilization | Increases HOMO-LUMO gap | Small-gap systems; Metals |
Convergence Challenges and Solutions
SCF convergence problems frequently occur in specific chemical contexts, including systems with very small HOMO-LUMO gaps, d- and f-elements with localized open-shell configurations, transition state structures with dissociating bonds, and systems described with non-physical calculation setups [7]. This section addresses practical strategies for identifying and resolving convergence difficulties.
Diagnostic Framework and Troubleshooting
When encountering SCF convergence issues, a systematic diagnostic approach is essential:
- Verify System Realism: Ensure bond lengths, angles, and other structural parameters are physically reasonable [7]
- Check Spin Multiplicity: Confirm the correct spin multiplicity is used, with open-shell systems computed using spin-unrestricted formalisms [7]
- Assess Initial Guess Quality: Examine whether a better initial guess (e.g., from a fragment calculation or previous computation) is available [1]
- Monitor Convergence Behavior: Strongly fluctuating errors may indicate an electronic configuration far from any stationary point [7]
Advanced Convergence Techniques
For persistently challenging systems, several advanced techniques can be employed:
Electron Smearing: This approach simulates a finite electron temperature using fractional occupation numbers to distribute electrons over near-degenerate levels, particularly helpful for metallic systems or those with small HOMO-LUMO gaps [1] [7]. The smearing parameter should be kept as low as possible and potentially reduced through multiple restarts [7].
Mixing Parameter Adjustment: Most quantum chemistry packages allow control over the mixing of the Fock or density matrices between iterations. For problematic cases, reducing the mixing parameter (e.g., to 0.015 instead of the default 0.2-0.25) can stabilize convergence, albeit at the cost of slower progress [8] [7].
DIIS Parameter Tuning: Adjusting DIIS-specific parameters can significantly impact convergence:
- Increasing the number of DIIS expansion vectors (e.g., from default 10 to 25) enhances stability [7]
- Delaying the start of DIIS acceleration (increasing Cyc parameter) allows initial equilibration [7]
- Using separate error vectors for α and β spins can prevent false convergence in unrestricted calculations [6]
Alternative Solvers: When standard methods fail, switching to specialized algorithms like the Augmented Roothaan-Hall (ARH) method, which directly minimizes the total energy using a preconditioned conjugate-gradient approach, can be effective [7].
Convergence Criteria and Thresholds
Different computational packages provide various convergence thresholds, typically controlling the maximum acceptable change in density matrix elements, energy between iterations, or the DIIS error vector. The following table summarizes standard convergence criteria in the ORCA package:
Table 3: Standard SCF Convergence Criteria in ORCA [9]
| Criterion | Loose | Medium | Strong | Tight | VeryTight |
|---|---|---|---|---|---|
| TolE (Energy) | 1e-5 | 1e-6 | 3e-7 | 1e-8 | 1e-9 |
| TolMaxP (Max Density) | 1e-3 | 1e-5 | 3e-6 | 1e-7 | 1e-8 |
| TolRMSP (RMS Density) | 1e-4 | 1e-6 | 1e-7 | 5e-9 | 1e-9 |
| TolErr (DIIS Error) | 5e-4 | 1e-5 | 3e-6 | 5e-7 | 1e-8 |
| TolG (Orbital Gradient) | 1e-4 | 5e-5 | 2e-5 | 1e-5 | 2e-6 |
For most single-point energy calculations, "Strong" convergence criteria are sufficient, while geometry optimizations and frequency calculations typically require "Tight" criteria [6] [9]. It is crucial to ensure that the integral evaluation threshold is compatible with the SCF convergence criteria, typically 3-5 orders of magnitude tighter than the SCF convergence tolerance [6].
Practical Protocols and Research Reagent Solutions
Step-by-Step Protocol for Challenging SCF Calculations
For systems exhibiting SCF convergence difficulties, the following structured protocol is recommended:
Phase 1: Initial Assessment and Basic Adjustments
- Geometry Validation: Verify molecular structure using chemical knowledge and preliminary molecular mechanics calculations [7]
- Spin State Confirmation: Determine the correct spin multiplicity through experimental data or preliminary calculations [7]
- Initial Guess Selection: Begin with 'minao' or 'atom' guess; for transition metals, try 'huckel' or atomic calculations [1]
- Basis Set Evaluation: Ensure the basis set is appropriate for the elements and properties of interest
Phase 2: Algorithm Selection and Parameter Optimization
- Start with Default DIIS: Use DIIS with default parameters for initial attempts [6]
- Adjust Mixing Parameters: If oscillations occur, reduce mixing weight to 0.1-0.15 [8] [7]
- Implement Damping: Apply damping (factor 0.5) for the first few iterations before DIIS acceleration begins [1]
- Consider Level Shifting: For small-gap systems, implement level shifting (0.001-0.01 Hartree) [1] [7]
Phase 3: Advanced Techniques for Resistant Cases
- Switch Algorithm: If DIIS fails after 30-50 cycles, switch to GDM or SOSCF [1] [6]
- Employ Smearing: Apply minimal electron smearing (0.001-0.005 Hartree) and gradually reduce it over multiple restarts [7]
- Fragment Initialization: For large systems, initialize calculation using converged densities of molecular fragments [1]
- Staged Convergence: Use successively tighter convergence criteria through restart procedures [1]
Phase 4: Post-Convergence Validation
- Stability Analysis: Perform SCF stability analysis to verify the solution is a true minimum [1] [9]
- Property Consistency: Check that calculated properties (dipole moments, populations) are chemically reasonable [1]
- Wavefunction Analysis: Examine molecular orbitals and orbital energies for physical consistency
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Computational Tools for SCF Research
| Tool Category | Specific Implementation | Function | Application Context |
|---|---|---|---|
| Initial Guess Methods | 'minao', 'atom', 'huckel' | Generate starting electron density | Default: 'minao'; Metals: 'huckel' |
| Convergence Accelerators | DIIS, GDM, SOSCF | Extrapolate to self-consistency | Standard: DIIS; Difficult: GDM/SOSCF |
| Stabilization Techniques | Damping, Level Shifting | Suppress oscillations | Initial cycles; Small-gap systems |
| Fractional Occupancy | Fermi smearing, Gaussian smearing | Treat near-degenerate states | Metallic systems; Small HOMO-LUMO gaps |
| Analysis Tools | Stability analysis, Population analysis | Validate solution quality | Post-convergence verification |
| Basis Sets | cc-pVTZ, cc-pVQZ, def2-TZVP | Expand molecular orbitals | Balance between accuracy and cost |
Stability Analysis and Validation
Even when an SCF calculation converges, the resulting wavefunction may not represent the true ground state. SCF solutions can correspond to saddle points rather than minima on the energy surface [1]. Stability analysis provides a critical validation step by examining whether the energy can be lowered by perturbing the orbitals [1].
Instabilities are conventionally classified as either internal or external [1]. Internal instabilities indicate that the calculation has converged to an excited state rather than the ground state, while external instabilities signify that energy could be lowered by relaxing constraints on the wavefunction, such as allowing restricted orbitals to become unrestricted [1]. Most quantum chemistry packages include tools for performing stability analysis, which should be routinely employed, particularly for systems with known convergence difficulties or when investigating novel electronic structures.
SCF theory provides a unified framework encompassing both Hartree-Fock and Kohn-Sham Density Functional Theory, serving as the computational backbone of modern quantum chemistry and materials science. The iterative SCF process—beginning with an initial guess, progressing through Fock matrix construction and diagonalization, and employing convergence acceleration—represents a powerful approach to solving the quantum many-body problem. Despite the mathematical complexity of the underlying equations, robust algorithms exist for solving the forward problem of predicting properties from molecular structures [10].
The continuing evolution of SCF methodologies, including recent advances in deep-learning-powered density functionals [3], ensures that SCF theory remains a vibrant and developing field. For researchers pursuing a thesis on SCF self-consistent field iterative processes, numerous challenges and opportunities remain, particularly in addressing inverse problems—inferring structural or model parameters from observed physical properties [10]. The robust algorithmic framework outlined in this work provides both a foundation for practical computational work and a springboard for future methodological development.
The Self-Consistent Field (SCF) method represents the computational cornerstone of modern quantum chemistry, enabling the determination of molecular orbitals and electronic energies within Hartree-Fock theory and Kohn-Sham Density Functional Theory. This iterative procedure solves a nonlinear eigenvalue problem where the Fock matrix (F) depends on its own solution, requiring self-consistent convergence [1]. The fundamental SCF equation in the atomic orbital (AO) basis takes the form of a generalized eigenvalue problem: F C = S C ε, where C is the matrix of molecular orbital coefficients, ε is a diagonal matrix of orbital energies, and S is the AO-basis overlap matrix [11]. The solution to this equation provides both the molecular orbitals that describe electron distribution and their associated energies, which are crucial for understanding molecular reactivity, stability, and properties.
The Fock matrix F serves as an effective one-electron potential that approximates the average field experienced by an electron due to the presence of all other electrons in the system. For a closed-shell system, the Fock matrix elements are constructed as F{αβ} = *h*{αβ} + ∑{γδ} *D*{γδ} [ ⟨αγ|βδ⟩ - ⟨αγ|δβ⟩ ], where h is the core Hamiltonian matrix, D is the density matrix, and the two-electron integrals represent electron repulsion [11]. This formulation captures the essential quantum mechanical interactions while maintaining computational tractability through the mean-field approximation. The SCF procedure's importance extends across multiple disciplines, from drug discovery where it enables protein-ligand binding energy calculations, to materials science where it facilitates the design of novel functional materials with tailored electronic properties.
Theoretical Foundation of the Fock Matrix
Wavefunction Approximation and Energy Expression
In Hartree-Fock theory, the N-electron wavefunction is approximated as a single Slater determinant |Ψ₀⟩, an antisymmetrized product of N one-electron functions called molecular orbitals (MOs) [12]. For a system with N electrons, the Slater determinant is constructed as:
where ψi(𝑥⃗) are the molecular orbitals, each being a function of the spatial and spin coordinates of a single electron [13]. The molecular orbitals are themselves expanded as a linear combination of atomic orbital (LCAO) basis functions χμ: ψi = ∑μ C{μi} χμ, where C{μi} are the molecular orbital coefficients [12]. The electronic energy within this approximation is given by EHF = ⟨Ψ₀|Ĥ|Ψ₀⟩ = ∑i ⟨i|ħ|i⟩ + ½ ∑{i,j} [⟨ii|jj⟩ - ⟨ij|ji⟩], where ħ is the one-electron core Hamiltonian, and the two-electron integrals represent electron repulsion [13].
The Hartree-Fock energy can be equivalently expressed in the AO basis as EHF = D{μν}^α (H{μν} + F{μν}^α) + D{μν}^β (H{μν} + F_{μν}^β), where D^α and D^β are the alpha and beta density matrices, H is the AO-basis one-electron Hamiltonian, and F^α and F^β are the Fock matrices for alpha and beta spins [13]. This formulation facilitates computational implementation while maintaining the theoretical rigor of the quantum mechanical treatment.
Variational Derivation and the SCF Equation
The optimal molecular orbitals are determined by minimizing the electronic energy with respect to the orbital coefficients C_{μi}, subject to the constraint that the orbitals remain orthonormal [12]. Applying the method of Lagrange multipliers to this constrained optimization problem leads to the Roothaan-Hall equations F C = S C ε for closed-shell systems [13]. Here, the Fock matrix F acts as the effective Hamiltonian, the overlap matrix S ensures proper normalization in non-orthogonal basis sets, C contains the orbital coefficients, and ε is a diagonal matrix of orbital energies that correspond to the Lagrange multipliers.
The Fock matrix depends on the density matrix D, which in turn is constructed from the occupied orbital coefficients: D{γδ} = ∑{i=1}^N C{γi}^* C{δi} for a general molecular orbital coefficient matrix, or D{μν} = C{μ i} C_{ν i} for real-valued orbitals in the AO basis [11] [13]. For a closed-shell system, the summation includes all doubly occupied orbitals. This interdependence makes the Hartree-Fock equation nonlinear and necessitates an iterative solution approach where an initial guess is progressively refined until self-consistency is achieved.
Mathematical Decomposition of the Fock Matrix
The Fock matrix can be decomposed into four fundamental components that represent distinct physical interactions within the molecular system: F = T + V + J + K [1]. Each component captures specific quantum mechanical effects and contributes uniquely to the total electronic energy and molecular orbital structure.
Table 1: Core Components of the Fock Matrix
| Component | Matrix Element | Physical Interpretation | Mathematical Properties | ||
|---|---|---|---|---|---|
| T (Kinetic Energy) | T_{μν} = ⟨μ | -½∇² | ν⟩ | Kinetic energy of electrons | Positive definite, spin-independent |
| V (Nuclear Attraction) | V_{μν} = ⟨μ | ∑A -ZA/r_{1A} | ν⟩ | Electron-nuclear Coulomb attraction | Negative definite, spin-independent |
| J (Coulomb) | J{μν} = ∑{γδ} D_{γδ} (μν | γδ) | Electron-electron repulsion | Depends on total density D, positive definite | |
| K (Exchange) | K{μν} = -½ ∑{γδ} D_{γδ} (μγ | δν) | Quantum mechanical exchange | Non-local, depends on spin case, negative contribution |
One-Electron Components: T and V
The kinetic energy matrix T represents the quantum mechanical kinetic energy of electrons and is constructed from integrals over the basis functions with the kinetic energy operator: T{μν} = ⟨χμ| -½∇² |χ_ν⟩ [12]. This matrix is always positive definite as kinetic energy is inherently positive, and it is independent of the electronic spin. The kinetic energy integrals are typically evaluated analytically for Gaussian-type basis functions, which are the standard in quantum chemistry.
The nuclear attraction matrix V captures the Coulomb attraction between electrons and atomic nuclei: V{μν} = ⟨χμ| ∑A -ZA/r{1A} |χν⟩, where ZA is the nuclear charge of atom A, and r{1A} is the distance between the electron and nucleus A [12]. This matrix is always negative definite since the electron-nuclear interaction is attractive. Together, T and V form the core Hamiltonian h = T + V, which represents the energy of electrons in the absence of electron-electron repulsion and serves as the initial guess in many SCF procedures [1].
Two-Electron Components: J and K
The Coulomb matrix J represents the classical electrostatic repulsion between electrons and is constructed as J{μν} = ∑{γδ} D{γδ} (μν|γδ), where (μν|γδ) are the two-electron repulsion integrals (ERIs) in chemists' notation: (μν|γδ) = ∬ χμ(𝑟⃗₁) χν(𝑟⃗₁) (1/r₁₂) χγ(𝑟⃗₂) χδ(𝑟⃗₂) d³𝑟⃗₁ d³𝑟⃗₂ [13] [12]. These integrals describe the Coulomb interaction between two charge distributions: one centered at χμχν and another at χγχ_δ. The J matrix depends on the total electron density through D and contributes positively to both the Fock matrix and the total energy.
The exchange matrix K arises from the antisymmetric nature of the wavefunction and the Pauli exclusion principle, with elements K{μν} = -½ ∑{γδ} D_{γδ} (μγ|δν) [13]. The exchange interaction is a purely quantum mechanical phenomenon with no classical analogue, representing the favorable energy lowering when electrons with parallel spins avoid each other. For restricted closed-shell Hartree-Fock, the prefactor ensures proper normalization, while the specific form changes for different spin cases in open-shell systems. The K matrix is non-local, meaning that its elements connect basis functions that may be spatially distant, though the magnitude decreases with separation.
Computational Implementation of SCF Methodology
The SCF Iterative Procedure
The SCF process follows a well-defined iterative workflow to achieve self-consistency between the Fock matrix and the molecular orbitals. The following diagram illustrates this iterative procedure:
The SCF cycle begins with an initial guess for the molecular orbitals or density matrix. Common initial guesses include the superposition of atomic densities (SAD) [13], the core Hamiltonian (ignoring electron-electron interactions) [1], or orbitals from a previous calculation [14]. The quality of this initial guess significantly impacts convergence behavior, with better guesses typically leading to faster convergence.
Once an initial guess is available, the iterative process proceeds through these steps:
- Form Density Matrix: Construct the density matrix D from the occupied molecular orbitals: D{γδ} = ∑{i=1}^N C{γ i} C{δ i} for a real-valued, closed-shell system [11].
- Build Fock Matrix: Compute the Fock matrix F = h + J - K, where h is the core Hamiltonian, and J and K are constructed using the current density matrix [12].
- Solve Roothaan-Hall Equation: Solve the generalized eigenvalue problem F C = S C ε to obtain updated orbitals and energies [11].
- Check Convergence: Assess whether the solution has converged based on changes in energy, density matrix, or the DIIS error vector [9].
This process repeats until convergence criteria are satisfied, indicating that a self-consistent solution has been reached.
Integral Evaluation and Technical Considerations
The computation of two-electron repulsion integrals (ERIs) represents the most computationally demanding aspect of Fock matrix construction. These integrals (μν|λσ) = ∬ χμ(𝑟⃗₁) χν(𝑟⃗₁) (1/r₁₂) χλ(𝑟⃗₂) χσ(𝑟⃗₂) d³𝑟⃗₁ d³𝑟⃗₂ must be evaluated for all combinations of basis functions [12]. Quantum chemistry packages employ various strategies for handling these integrals:
- Conventional SCF: Store all ERIs on disk and read them each iteration [15]
- Direct SCF: Recompute integrals as needed each iteration [15]
- Density Fitting (DF): Approximate ERIs using three-index integrals to reduce computational cost and storage requirements [12]
Density fitting, also known as the resolution-of-the-identity approximation, represents a particularly efficient approach where the four-index ERIs are approximated using three-index integrals: (μν|λσ) ≈ ∑_Q (μν|Q) (Q|λσ), where Q indexes auxiliary basis functions [12]. This reduces the formal scaling of memory requirements from O(N⁴) to O(N³) and computational cost from O(N⁴) to O(N³), though the constant factors and practical performance depend on implementation details.
Basis Sets and Their Role in SCF Calculations
The choice of basis set {χ_μ} fundamentally determines the accuracy and computational cost of SCF calculations. Basis functions are typically contracted Cartesian Gaussian functions centered on atomic nuclei [13]. Common basis set types include:
- Minimal Basis Sets: Contain the minimum number of functions needed for each atom (e.g., STO-3G)
- Double-Zeta Basis Sets: Include two functions for each valence orbital (e.g., cc-pVDZ)
- Triple-Zeta Basis Sets: Provide three functions for each valence orbital (e.g., cc-pVTZ)
- Polarized Basis Sets: Add higher angular momentum functions for flexibility (e.g., cc-pVXZ series)
- Diffuse Functions: Include spatially extended functions for better description of anions and excited states
The overlap matrix S with elements S{μν} = ⟨χμ|χ_ν⟩ accounts for non-orthogonality between basis functions [12]. During the SCF procedure, the generalized eigenvalue problem is often transformed to an orthogonal basis using Löwdin's symmetric orthogonalization, which employs the transformation matrix X = S^{-1/2} [11] [12].
Convergence Acceleration and Stability Analysis
Convergence Criteria and Thresholds
SCF convergence is typically assessed using multiple criteria that monitor changes in key quantities between iterations. Different quantum chemistry packages implement various convergence thresholds, as illustrated by ORCA's comprehensive set of criteria:
Table 2: SCF Convergence Criteria in ORCA [9]
| Criterion | TightSCF Value | Physical Meaning | Role in Convergence |
|---|---|---|---|
| TolE | 1e-8 | Energy change between cycles | Ensures energy stability |
| TolRMSP | 5e-9 | RMS density matrix change | Measures matrix self-consistency |
| TolMaxP | 1e-7 | Maximum density matrix change | Captures worst-case element change |
| TolErr | 5e-7 | DIIS error vector norm | Quantifies commutator [F,P] |
| TolG | 1e-5 | Orbital gradient convergence | Ensures proper energy minimum |
These criteria ensure that the SCF procedure terminates only when the solution has reached sufficient numerical stability. The ConvCheckMode parameter determines how strictly these criteria are applied, with ConvCheckMode=0 requiring all criteria to be satisfied, while ConvCheckMode=2 provides a balanced approach checking both total and one-electron energy changes [9].
Convergence Acceleration Algorithms
Several algorithms exist to improve SCF convergence, particularly for challenging systems with small HOMO-LUMO gaps or complex electronic structures:
- DIIS (Direct Inversion in Iterative Subspace): Extrapolates the Fock matrix using information from previous iterations by minimizing the norm of the commutator [F, D S] [1] [15]. DIIS is the default in most quantum chemistry packages due to its effectiveness for most systems.
- EDIIS (Energy-DIIS): Combines energy interpolation with DIIS for improved stability [1].
- ADIIS (Augmented DIIS): An alternative DIIS variant that can be more robust for difficult cases [1].
- Damping: Mixes a fraction of the previous Fock matrix with the new one: F{new} = λ F{old} + (1-λ) F_{new}, typically with λ = 0.5 or smaller [1].
- Level Shifting: Artificially increases the energy of virtual orbitals to improve convergence stability [1] [7].
- Fermi Smearing: Uses fractional occupancies based on a temperature function to facilitate convergence in metallic systems or those with small gaps [1] [7].
- Quadratic Convergent (QC) SCF: Employs second-order convergence algorithms that guarantee convergence close to the solution but with higher computational cost per iteration [15].
For open-shell systems, additional complications arise, and methods like Average-of-Configuration (AOC) or Fractional Occupation (FOCC) approaches may be employed [14]. The AOC method is the default for open-shell Hartree-Fock in DIRAC, while FOCC is preferred for Kohn-Sham calculations [14].
Stability Analysis
Even after SCF convergence, the resulting wavefunction may represent a saddle point rather than a true minimum [1]. Stability analysis checks whether the solution corresponds to a local minimum by examining the orbital rotation Hessian matrix. Instabilities are classified as:
- Internal Instabilities: The solution corresponds to an excited state within the same symmetry and spin constraints
- External Instabilities: Lower energy exists with different symmetry (e.g., restricted → unrestricted) or spatial symmetry breaking
When instabilities are detected, the calculation can be restarted with modified constraints or initial guesses to locate the true ground state [1].
Research Reagent Solutions: Computational Tools for SCF Development
Table 3: Essential Computational Tools for SCF Method Development
| Tool/Category | Example Implementations | Primary Function | Application Context |
|---|---|---|---|
| Integral Evaluation | Psi4 MintsHelper, Libint | Compute 1e- and 2e- integrals | Fock matrix construction |
| Linear Algebra | BLAS, LAPACK, Eigen | Matrix operations, diagonalization | Solve F C = S C ε |
| Density Fitting | Psi4 DFTensor, DFCOEF files | Approximate 4-index ERIs with 3-index | Reduce computational cost |
| DIIS Extrapolation | PySCF DIIS, EDIIS, ADIIS | Accelerate SCF convergence | Improve iteration efficiency |
| Basis Set Libraries | Basis Set Exchange, internal libs | Provide atomic orbital basis sets | Molecular representation |
These computational tools form the essential "reagent kit" for implementing and advancing SCF methodologies. The MintsHelper class in Psi4 provides access to one-electron integrals including kinetic energy (T), nuclear attraction (V), and overlap (S) matrices [12]. The DFTensor class handles density-fitted two-electron integrals, which are crucial for efficient Fock matrix construction in large systems [12]. BLAS and LAPACK libraries provide optimized linear algebra routines for matrix operations and diagonalization, which are performance-critical for solving the Roothaan-Hall equations.
Advanced Applications and Specialized Formulations
Open-Shell Systems and Spin Treatment
The fundamental Fock matrix decomposition extends to open-shell systems through several specialized formulations:
- Unrestricted Hartree-Fock (UHF): Employs separate spatial orbitals for α and β spins, with Fock matrices F^α and F^β that depend on both D^α and D^β [13]. This approach provides maximum flexibility but may suffer from spin contamination.
- Restricted Open-Shell Hartree-Fock (ROHF): Maintains double occupation of closed-shell orbitals while allowing open-shell orbitals with constrained coefficients [13]. This preserves spin purity but offers less variational flexibility.
- Average-of-Configuration (AOC): Treats open shells using fractionally occupied orbitals to produce state-averaged solutions, particularly useful for degenerate systems [14].
For the UHF case, the Fock matrices for α and β spins are constructed as F^α = h + J - K^α and F^β = h + J - K^β, where J depends on the total density D = D^α + D^β, while the exchange components K^α and K^β depend only on like-spin densities [13].
Density Functional Theory Integration
In Kohn-Sham Density Functional Theory (KS-DFT), the Fock matrix is augmented with an exchange-correlation term: FKS = h + J - K + VXC [1]. The exchange-correlation potential V_XC is typically evaluated numerically on a grid and incorporates electron correlation effects missing in Hartree-Fock theory. The SCF procedure remains essentially unchanged, though convergence characteristics may differ due to the properties of the specific functional employed.
Linear Scaling and High-Performance Approaches
For large systems containing thousands of atoms, traditional O(N³) or O(N⁴) scaling becomes prohibitive. Linear-scaling SCF approaches exploit the sparsity of matrices in large systems through:
- Sparse Matrix Algebra: Exploiting distance-based screening to neglect small matrix elements
- Domain Decomposition: Partitioning the system into localized regions
- Incremental Fock Builds: Updating only significantly changed portions of the Fock matrix between iterations [16]
- Continuous Fast Multipole Method: Efficient evaluation of long-range Coulomb interactions
These approaches enable SCF calculations on systems of unprecedented size, bridging the gap between molecular quantum chemistry and materials simulation.
The decomposition of the Fock matrix into its fundamental components F = T + V + J + K provides both conceptual clarity and computational efficiency for solving the electronic structure problem. The kinetic energy T and nuclear attraction V matrices capture one-electron effects, while the Coulomb J and exchange K matrices describe electron-electron interactions within the mean-field approximation. Through iterative solution of the SCF equations, these components self-consistently determine molecular orbitals and energies that form the foundation for predicting molecular structure, reactivity, and properties.
Advancements in algorithm development, including sophisticated convergence acceleration techniques, density fitting approximations, and linear-scaling approaches, continue to expand the applicability of SCF methods to increasingly complex systems. As computational resources grow and algorithms improve, the detailed understanding of Fock matrix components remains essential for both method development and practical application across chemistry, materials science, and drug discovery.
The method of Linear Combination of Atomic Orbitals (LCAO) represents a fundamental approximation in quantum chemistry for calculating molecular orbitals. First introduced by Sir John Lennard-Jones in 1929 with descriptions of bonding in diatomic molecules, this technique employs a quantum superposition of atomic orbitals to construct molecular orbitals [17]. In practical computational chemistry, the LCAO approach transforms the complex integro-differential equations of quantum mechanics into tractable algebraic equations suitable for numerical implementation on computers [18].
In the LCAO formalism, each molecular orbital φi* is expressed as a linear expansion of atomic basis functions χμ* according to the equation: $$ \phii = \sum{\mu} c{\mu i} \chi{\mu} $$ where cμi* represents the molecular orbital coefficients that determine the contribution of each atomic orbital to the molecular orbital [19] [17]. The number of molecular orbitals obtained exactly equals the number of atomic orbitals included in the linear expansion, creating a one-to-one correspondence between basis functions and resulting molecular orbitals [17]. This expansion technique is central to most modern electronic structure calculations, including Hartree-Fock theory and density functional theory, where it provides the mathematical framework for solving the electronic Schrödinger equation for molecular systems [20].
Mathematical Foundation of the LCAO Method
The Roothaan-Hall Equations
The application of the LCAO approximation within the Hartree-Fock method leads to the Roothaan-Hall equations, which form the cornerstone for computational quantum chemistry. These matrix equations convert the complex integro-differential Hartree-Fock equations into a generalized eigenvalue problem of the form [19] [21]: $$ \mathbf{FC} = \mathbf{SC\epsilon} $$ Here, F represents the Fock matrix, C is a square matrix containing the molecular orbital coefficients, S denotes the atomic orbital overlap matrix with elements Sμν* = ⟨χμ|χν⟩, and ε is a diagonal matrix of the orbital energies [21]. The Fock matrix elements incorporate both one-electron and two-electron contributions and can be expressed as: $$ F{\mu,\nu} = \langle \chi\mu | h | \chi\nu \rangle + \sum{\delta,\kappa} \left[ \gamma{\delta,\kappa} \langle \chi\mu \chi\delta | g | \chi\nu \chi\kappa \rangle - \gamma{\delta,\kappa}^{ex} \langle \chi\mu \chi\delta | g | \chi\kappa \chi\nu \rangle \right] $$ where γδ,κ* represents the density matrix elements [19].
For systems with unpaired electrons, this formalism extends to the Pople-Nesbet equations, which consist of two coupled matrix equations for alpha and beta spins [21]: $$ \mathbf{F^\alpha C^\alpha} = \mathbf{SC^\alpha \epsilon^\alpha} $$ $$ \mathbf{F^\beta C^\beta} = \mathbf{SC^\beta \epsilon^\beta} $$ These equations maintain the same structural form as the Roothaan-Hall equations but accommodate different spatial distributions for spin-up and spin-down electrons [21].
Density Matrix Formulation
Within the LCAO framework, the electron density plays a pivotal role in quantifying the probability of finding an electron at a specific position in space. The spin-σ electron density is computed as [20]: $$ \rho\sigma(\mathbf{r}) = \sum{i=1}^{N\sigma} \sum{\mu\nu} C{\mu i}^\sigma C{\nu i}^\sigma \chi\mu(\mathbf{r}) \chi\nu(\mathbf{r}) = \sum{\mu\nu} P{\mu\nu}^\sigma \chi\mu(\mathbf{r}) \chi\nu(\mathbf{r}) $$ Here, Pμνσ represents the density matrix elements for spin σ, defined as $P{\mu\nu}^\sigma = \sum{i=1}^{N\sigma} C{\mu i}^\sigma C_{\nu i}^\sigma$ [20]. The total electron density is obtained by summing the contributions from both spin densities: ρ(r) = ρα(r) + *ρβ*(r) [20]. This density matrix formulation provides the crucial link between the molecular orbital coefficients and the observable electron distribution in molecules.
The Self-Consistent Field Iterative Procedure
Theoretical Framework of SCF Convergence
The Self-Consistent Field (SCF) method constitutes an iterative computational procedure for solving the quantum chemical equations derived from the LCAO approximation. The fundamental structure of the SCF process can be represented as a fixed-point problem where the electron density ρ is determined through repeated application of the SCF step function F according to ρ = D(V(ρ)) [22]. In this formulation, V represents the potential dependent on the density ρ, while D signifies the potential-to-density mapping, which involves constructing the Hamiltonian and diagonalizing it to obtain new eigenpairs [22].
The convergence behavior of SCF algorithms near the fixed point ρ* can be analyzed by examining the error propagation between iterations. Considering the error at iteration n as en* = ρn* - ρ*, the error in subsequent iterations follows the relationship [22]: $$ e{n+1} \simeq [1 - \alpha P^{-1} \varepsilon^\dagger] en = [1 - \alpha P^{-1} \varepsilon^\dagger]^n e_0 $$ Here, α represents a damping parameter, P−1 denotes a preconditioner, and ε† = [1 - χ0K] defines the dielectric matrix adjoint operator, which incorporates the independent-particle susceptibility χ0 and the Hartree-exchange-correlation kernel K [22]. The convergence of the SCF procedure requires that all eigenvalues of the matrix [1 - αP−1ε†] lie between -1 and 1 [22].
SCF Algorithm and Convergence Optimization
The practical implementation of the SCF method follows a well-defined iterative cycle, as illustrated below:
Diagram 1: The SCF iterative procedure, showing the cyclic process of density update and convergence checking.
The convergence efficiency of SCF calculations depends critically on two factors: the damping parameter α and the preconditioner P−1. The optimal damping parameter is given by: $$ \alpha{opt} = \frac{2}{\lambda{\min} + \lambda_{\max}} $$ where λmin* and λmax* represent the smallest and largest eigenvalues of P−1ε†, respectively [22]. The rate of convergence correlates with the spectral condition number κ = λmax/λmin, with smaller values leading to faster convergence [22]. For challenging systems, advanced initial guess strategies like Basis Set Projection (BSP) and Many-Body Expansion (MBE) can reduce total computational wall-time by up to 27.6% compared to conventional superposition of atomic densities approaches [23].
Basis Sets in LCAO Calculations
Types of Basis Functions
The choice of basis functions represents a critical consideration in LCAO calculations, with two primary types dominating computational quantum chemistry:
Table 1: Comparison of Primary Basis Function Types
| Basis Type | Mathematical Form | Advantages | Limitations |
|---|---|---|---|
| Slater-Type Orbitals (STO) | $χ{n,l,m}(r,θ,φ) = N{n,l,m,ξ} Y_{l,m}(θ,φ) r^{n-1} e^{-ξr}$ [24] | Correct electron-nuclear cusp behavior; accurate decay far from nucleus [18] [24] | Computational complexity for polyatomic molecules [24] |
| Gaussian-Type Orbitals (GTO) | $χ{a,b,c}(r,θ,φ) = N'{a,b,c,α} x^a y^b z^c e^{-αr^2}$ [24] | Efficient computation of multi-center integrals [18] [24] | Approximate representation requiring more functions [18] |
The key advantage of GTOs lies in their computational efficiency for molecular calculations. When multiplying two Gaussian functions centered at different positions, the product can be expressed as a single Gaussian centered at an intermediate point through the relation: $$ e^{-\alphaa (\mathbf{r}-\mathbf{R}a)^2} e^{-\alphac (\mathbf{r}-\mathbf{R}c)^2} = e^{-(\alphaa + \alphac)(\mathbf{r}-\mathbf{R}')^2} e^{-\alpha' (\mathbf{R}a-\mathbf{R}c)^2} $$ where $\mathbf{R}' = \frac{\alphaa\mathbf{R}a + \alphac\mathbf{R}c}{\alphaa + \alphac}$ and $\alpha' = \frac{\alphaa \alphac}{\alphaa + \alphac}$ [24]. This unique mathematical property enables efficient computation of the multi-center integrals that arise in polyatomic molecular calculations, establishing GTOs as the predominant choice in modern computational chemistry [24].
Basis Set Classification and Hierarchy
Basis sets are systematically organized into hierarchies that balance accuracy and computational cost:
Table 2: Classification of Gaussian Basis Sets
| Basis Set Type | Description | Examples | Typical Applications |
|---|---|---|---|
| Minimal | Single basis function for each atomic orbital | STO-3G, STO-4G [18] | Preliminary calculations for large systems |
| Split-Valence | Multiple functions for valence orbitals | 3-21G, 6-31G, 6-311G [18] | Standard molecular calculations |
| Polarized | Added angular momentum functions | 6-31G, 6-31G* [18] | Accurate geometry optimization |
| Diffuse | Additional functions with small exponents | 6-31+G, 6-311++G [18] | Anions, excited states, weak interactions |
| Correlation-Consistent | Systematic path to complete basis set limit | cc-pVDZ, cc-pVTZ, cc-pVQZ [18] [25] | High-accuracy correlated calculations |
The Pople-style notation X-YZG conveys specific information about the basis set composition: X indicates the number of primitive Gaussians forming each core atomic orbital basis function, while Y and Z specify that valence orbitals are composed of two basis functions, each composed of linear combinations of Y and Z primitive Gaussian functions, respectively [18]. Additional notations include "*" for polarization functions on heavy atoms and "+*" for diffuse functions on heavy atoms [18].
For high-accuracy calculations, particularly those incorporating electron correlation effects, correlation-consistent basis sets developed by Dunning and coworkers provide a systematic pathway to approach the complete basis set (CBS) limit [18] [25]. These basis sets, designated as cc-pVNZ where N = D (double), T (triple), Q (quadruple), 5, 6, etc., are specifically designed for smooth convergence in post-Hartree-Fock calculations [18]. Recent developments have extended these principles to periodic systems, generating specialized Gaussian basis sets for solids that maintain numerical stability while providing smooth convergence to the CBS limit [25].
Computational Implementation and Protocols
SCF Algorithm Implementation
The practical implementation of SCF algorithms requires careful attention to numerical procedures and convergence criteria. A basic implementation of the fixed-point iteration algorithm can be structured as follows [22]:
- Initialization: Begin with an initial guess density ρ0 and associated metadata
- Iteration Cycle: For n = 1 to maximum iterations:
- Compute the Fock matrix F(ρn*)
- Solve the generalized eigenvalue problem FC = SCε
- Construct new density ρn+1 from the molecular orbitals
- Check convergence criterion ||ρn+1 - ρn|| < *δ
- If converged, exit loop; else continue
- Termination: Return the converged density and associated energy
For production-level calculations, more sophisticated convergence acceleration techniques are employed, including density mixing schemes of the form: $$ \rho{n+1} = \rhon + \alpha P^{-1} (D(V(\rhon)) - \rhon) $$ where α represents a damping parameter and P−1 denotes a preconditioner designed to approximate the inverse dielectric operator [22].
Research Reagent Solutions: Computational Tools
Table 3: Essential Computational Components in LCAO-SCF Calculations
| Component | Function | Implementation Examples |
|---|---|---|
| Basis Set Library | Provides atomic orbital basis functions | Standardized Gaussian basis sets (cc-pVNZ, 6-31G*, etc.) [18] |
| Integral Evaluation | Computes molecular integrals over basis functions | Algorithms for one-electron and two-electron integrals [24] |
| Diagonalization Solver | Solves FC = SCε eigenvalue problem | Direct diagonalization, iterative methods [22] |
| Density Mixing | Accelerates SCF convergence | Simple mixing, Pulay DIIS, energy-derived preconditioners [22] |
| Initial Guess Methods | Provides starting electron density | Superposition of Atomic Densities (SAD), Basis Set Projection (BSP), Many-Body Expansion (MBE) [23] |
The relationships between these computational components and the overall SCF workflow can be visualized as:
Diagram 2: Dependency relationships between computational components in LCAO-SCF calculations.
Advanced initial guess methods have demonstrated significant improvements in computational efficiency. Recent research shows that Basis Set Projection (BSP) and Many-Body Expansion (MBE) techniques can reduce total wall-time by up to 21.9-27.6% for Hartree-Fock and hybrid density functional theory calculations on systems containing up to 14,386 basis functions compared to conventional superposition of atomic densities approaches [23]. A hybrid MBE-BSP method further enhances performance for challenging systems, though some convergence challenges remain for triplet electronic states [23].
The LCAO approach provides the fundamental mathematical framework for modern computational quantum chemistry, enabling the transformation of continuous quantum mechanical equations into discrete algebraic problems amenable to numerical computation. When combined with the SCF iterative procedure, this methodology allows for the practical determination of molecular electronic structure across diverse chemical systems. The careful selection of basis sets—from minimal to correlation-consistent—establishes a balance between computational efficiency and accuracy, while advanced algorithms for integral computation, matrix diagonalization, and convergence acceleration ensure robust performance. Ongoing developments in basis set design, initial guess methodologies, and SCF algorithms continue to expand the applicability of these techniques to increasingly complex molecular systems and materials, maintaining the LCAO-SCF approach as an indispensable tool in theoretical chemistry and computational drug discovery.
In quantum chemistry and condensed matter physics, determining the electronic structure of many-body systems represents a fundamental challenge. The self-consistent field (SCF) method provides a practical framework for approximating solutions to the many-electron Schrödinger equation by employing a mean-field approximation [4]. This approach transforms the intractable many-body problem into an effective one-body problem where each electron experiences the average field created by all other electrons, rather than their instantaneous interactions [26]. The SCF method is foundational to both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (DFT), serving as the computational backbone for most electronic structure calculations in chemistry and materials science [2] [1].
Within the SCF framework, the mean-field approximation offers computational tractability at the cost of simplifying electron-electron interactions. This simplification inherently neglects electronic correlation—the correlated movement of electrons that arises from their mutual Coulomb repulsion [4]. While the Hartree-Fock method fully accounts for fermionic exchange correlation through the antisymmetry of the wavefunction, it completely neglects Coulomb correlation, leading to systematic errors in predicted properties [4]. Understanding the physical interpretation of this approximation, its limitations, and advanced methods that transcend the mean-field picture is essential for researchers applying these computational techniques to problems in drug development and materials design.
Theoretical Foundation of the Mean-Field Approximation
The Born-Oppenheimer and Independent Electron Approximations
The SCF method rests on several key approximations that enable practical computation of electronic structure. The Born-Oppenheimer approximation separates electronic and nuclear motion, treating nuclei as fixed particles within the electronic Hamiltonian [4]. This approximation is justified by the significant mass difference between electrons and nuclei, allowing electrons to instantaneously adjust to nuclear positions.
The central approximation in mean-field theories is the representation of the many-electron wavefunction as a single Slater determinant—an antisymmetrized product of one-electron wavefunctions (orbitals) [4]. This formulation naturally incorporates the Pauli exclusion principle through the antisymmetry of the determinant, ensuring that no two electrons occupy the same quantum state. Mathematically, for an N-electron system, the wavefunction is expressed as:
[ \Phi0 = \mathcal{A}|\psi1(1)\psi2(2) \ldots \psiN(N)| ]
where (\mathcal{A}) is the antisymmetrization operator, and (\psi_i) are the one-electron spin orbitals [1].
The Fock Operator and Self-Consistency
In the Hartree-Fock method, the electronic energy is minimized with respect to the orbitals, leading to the Fock operator, an effective one-electron Hamiltonian:
[ \mathbf{F} = \mathbf{T} + \mathbf{V} + \mathbf{J} + \mathbf{K} ]
where (\mathbf{T}) represents the kinetic energy operator, (\mathbf{V}) the external potential (typically electron-nuclear attraction), (\mathbf{J}) the Coulomb operator, and (\mathbf{K}) the exchange operator [1]. The Fock operator depends on the orbitals themselves through the (\mathbf{J}) and (\mathbf{K}) operators, creating a nonlinear problem that must be solved iteratively.
The self-consistency condition requires that the orbitals used to construct the Fock operator are the same as the eigenfunctions of that operator [4] [1]. This leads to the pseudoeigenvalue equation:
[ \mathbf{F} \mathbf{C} = \mathbf{S} \mathbf{C} \mathbf{E} ]
where (\mathbf{C}) is the matrix of molecular orbital coefficients, (\mathbf{S}) is the atomic orbital overlap matrix, and (\mathbf{E}) is a diagonal matrix of orbital energies [1]. This equation is solved iteratively through the SCF procedure until convergence is reached.
Table 1: Key Approximations in the Hartree-Fock Mean-Field Method
| Approximation | Physical Meaning | Consequence |
|---|---|---|
| Born-Oppenheimer | Separation of electronic and nuclear motion | Allows focus on electronic structure alone |
| Single Slater Determinant | Wavefunction as antisymmetrized product of orbitals | Neglects most electron correlation effects |
| Mean-Field Interaction | Each electron experiences average field of others | Neglects instantaneous electron-electron correlations |
| Non-Relativistic | Neglects relativistic effects | Limited accuracy for heavy elements |
| Finite Basis Set | Expansion of orbitals in limited basis set | Introduces basis set truncation error |
Electronic Correlation: The Missing Piece
Physical Interpretation of Electron Correlation
Electronic correlation refers to the fact that the motion of electrons in a many-body system is correlated due to their Coulomb repulsion, meaning that the probability of finding two electrons at specific positions is not simply the product of independent probabilities [4]. This correlation effect arises because electrons avoid each other more effectively than predicted by the mean-field approximation, leading to a reduction in the electron-electron repulsion energy.
In the Hartree-Fock method, the exchange term correctly models the fact that two electrons with the same spin cannot occupy the same position in space (Fermi hole), but it fails to account for the additional avoidance between electrons of opposite spin (Coulomb hole) [4]. This missing correlation energy, while typically small compared to the total energy (often <1%), can be chemically significant and crucial for predicting reaction barriers, binding energies, and electronic excited states.
Classification of Correlation Effects
Electron correlation can be categorized into several types:
Dynamical correlation: Short-range correlations arising from the instantaneous Coulomb repulsion between electrons. This represents the majority of the correlation energy and is relatively uniform in space.
Non-dynamical (static) correlation: Occurs in systems with near-degenerate electronic configurations where a single Slater determinant provides a poor zeroth-order description. This is particularly important in bond dissociation, transition metal complexes, and diradicals.
Long-range correlation: Includes dispersion interactions (van der Waals forces) that are completely absent in Hartree-Fock theory but critical for describing intermolecular interactions in drug design and supramolecular chemistry.
The mean-field approximation's failure to describe London dispersion forces represents one of its most significant limitations for pharmaceutical applications, where accurate modeling of intermolecular interactions is essential for predicting binding affinities [4].
Beyond Conventional Mean-Field: Advanced Approaches
Dynamical Mean-Field Theory (DMFT)
For strongly correlated materials where conventional density functional theory approximations fail, Dynamical Mean-Field Theory (DMFT) has emerged as a powerful approach that extends the mean-field concept while preserving local temporal fluctuations [27] [28]. DMFT maps the original lattice problem onto an effective quantum impurity model—a single atom or site coupled to a self-consistent electron bath [28]. This approach captures the crucial interplay between localization and itinerancy that characterizes strongly correlated systems.
The self-consistency condition in DMFT requires that the impurity Green's function equals the local lattice Green's function:
[ G{\mathrm{imp}}(i\omegan) = G{ii}(i\omegan) = \sumk \frac{1}{i\omegan + \mu - \epsilon(k) - \Sigma(k, i\omega_n)} ]
The key DMFT approximation assumes the lattice self-energy is momentum-independent but retains its frequency dependence: (\Sigma(k, i\omegan) \approx \Sigma{\mathrm{imp}}(i\omega_n)) [28]. This approximation becomes exact in the limit of infinite lattice coordination and provides a reasonable description for three-dimensional systems.
Diagram 1: The DMFT Self-Consistency Loop
Post-Hartree-Fock Methods
To address the correlation problem within the wavefunction framework, several post-Hartree-Fock methods have been developed:
Configuration Interaction (CI): Expands the wavefunction as a linear combination of Slater determinants representing excitations from the reference Hartree-Fock wavefunction.
Coupled Cluster (CC): Uses an exponential ansatz to generate a correlated wavefunction that size-consistently incorporates excitations to all orders.
Møller-Plesset Perturbation Theory: Treats electron correlation as a perturbation to the Hartree-Fock Hamiltonian.
These methods systematically improve upon the mean-field description but at significantly increased computational cost, limiting their application to small and medium-sized systems in drug development.
Density Functional Theory Approaches
Modern density functional theory incorporates electron correlation through the exchange-correlation functional, which in principle should capture all many-body effects [2]. Practical calculations employ various approximations:
- Local Density Approximation (LDA): Uses the correlation energy of a uniform electron gas.
- Generalized Gradient Approximation (GGA): Incorporates density gradients.
- Hybrid functionals: Mix Hartree-Fock exchange with DFT exchange-correlation.
The combined LDA+DMFT method represents a particularly powerful approach for strongly correlated materials, where conventional DFT fails dramatically, such as in Mott insulators and heavy-fermion systems [27].
Practical Implementation and Convergence
The SCF Iterative Cycle
The practical implementation of mean-field methods follows an iterative SCF procedure that cycles through several well-defined steps. This process begins with an initial guess for the density or wavefunction and proceeds until self-consistency is achieved [29] [1].
Diagram 2: The SCF Iterative Process
Convergence Acceleration Techniques
Achieving SCF convergence can be challenging, particularly for systems with small HOMO-LUMO gaps, metallic character, or complex electronic structures. Several acceleration and stabilization techniques are commonly employed [29] [1]:
Table 2: SCF Convergence Acceleration Methods
| Method | Principle | Applicability |
|---|---|---|
| DIIS (Direct Inversion in Iterative Subspace) | Extrapolates Fock matrix by minimizing error norm | Default method for most systems |
| Damping | Mixes old and new Fock matrices with fixed ratio | Stabilizes oscillatory convergence |
| Level Shifting | Increases energy gap between occupied and virtual orbitals | Helps convergence in small-gap systems |
| Broyden Mixing | Quasi-Newton scheme using approximate Jacobians | Metallic and magnetic systems |
| SOSCF (Second-Order SCF) | Uses orbital Hessian for quadratic convergence | Difficult cases with near-degeneracies |
Monitoring Convergence Criteria
SCF convergence is typically monitored using multiple criteria to ensure a fully self-consistent solution has been obtained [29]:
- Density matrix convergence: The change in density matrix elements between iterations should fall below a specified threshold (e.g., (10^{-4})).
- Energy convergence: The change in total electronic energy between iterations should be negligible.
- Hamiltonian convergence: The change in Fock or Hamiltonian matrix elements can be monitored.
In SIESTA, for example, both density matrix and Hamiltonian convergence criteria are typically enabled, with default tolerances of (10^{-4}) and (10^{-3}) eV, respectively [29].
Research Toolkit: Computational Methods for Electronic Structure
Table 3: Essential Computational Tools for Electronic Structure Research
| Method/Technique | Description | Correlation Treatment | Computational Scaling |
|---|---|---|---|
| Hartree-Fock (HF) | Prototypical mean-field method | Exchange only, no correlation | (N^3)-(N^4) |
| Density Functional Theory (DFT) | Uses electron density as fundamental variable | Approximate exchange-correlation functional | (N^2)-(N^3) |
| Dynamical Mean-Field Theory (DMFT) | Effective impurity model with dynamic mean field | Local, dynamic correlations | Exponential in basis size |
| Configuration Interaction (CI) | Multideterminantal wavefunction expansion | Systematic improvement possible | (N^6)-(N^8) |
| Coupled Cluster (CC) | Exponential ansatz for wavefunction | Gold standard for molecular systems | (N^6)-(N^7) |
| Quantum Monte Carlo (QMC) | Stochastic sampling of wavefunction | Potentially exact, limited by fixed-node error | (N^3)-(N^4) |
Applications in Pharmaceutical Research
The mean-field approximation and its correlated extensions find numerous applications in drug development and pharmaceutical research:
Protein-ligand binding affinity prediction: DFT calculations provide insights into interaction energies, though dispersion-corrected functionals are essential for accuracy.
Reaction mechanism elucidation: Studying enzymatic catalysis and drug metabolism pathways through transition state modeling.
Electronic excitation modeling: Time-dependent DFT calculations for predicting UV-Vis spectra of drug molecules.
Solvation effects: Continuum solvation models combined with quantum chemical calculations for predicting solubility and partitioning behavior.
For pharmaceutical applications, the choice of computational method involves careful balancing of accuracy and computational cost, with hybrid DFT methods often providing the best compromise for studying systems of biological relevance.
The mean-field approximation provides a powerful conceptual and computational framework for electronic structure theory, reducing the complex many-electron problem to a tractable form. While this approximation neglects electron correlation effects that can be chemically significant, it establishes the foundation upon which more accurate methods are built. Contemporary computational chemistry employs a multi-tiered approach, using mean-field methods for initial exploration and progressively more sophisticated correlation treatments for final accuracy. The ongoing development of methods like DMFT for strongly correlated systems and efficient post-Hartree-Fock approaches for molecular applications continues to expand the reach of quantum chemical calculations in pharmaceutical research and materials design. Understanding the physical interpretation of the mean-field approximation and its limitations remains essential for the appropriate application of these computational tools to challenging problems in drug development.
The Roothaan-Hall and Pople-Nesbet equations represent the cornerstone of modern computational quantum chemistry, providing the mathematical framework that enables the numerical solution of the Hartree-Fock equations for molecular systems. Before their development, Hartree-Fock theory existed as a set of near-intractable integro-differential equations that could only be solved numerically for atoms or one-center problems [30]. This limitation was profoundly overcome in 1951 when Clemens C. J. Roothaan and George G. Hall independently linearized the Hartree-Fock problem by expanding the molecular orbitals in a finite, fixed basis set, thus transforming the problem into a matrix formulation amenable to computational solution [31] [20]. This approach, generally called the Roothaan-Hall method, applies specifically to closed-shell molecules where all molecular orbitals are doubly occupied, within what is known as Restricted Hartree-Fock (RHF) theory [31].
Subsequently, the formalism was extended to open-shell systems where the number of alpha and beta electrons differs. This extension was independently developed by Pople, Nesbet, and Berthier, leading to the Pople-Nesbet-Berthier equations, which form the basis of Unrestricted Hartree-Fock (UHF) theory [32] [20]. In UHF theory, different spatial orbitals are used for alpha and beta electrons, described as a "different orbitals for different spins" (DODS) approach [32]. These developments collectively form the mathematical foundation for self-consistent field (SCF) procedures that are implemented in virtually all ab initio quantum chemistry programs today [30] [20]. The significance of these equations lies in their transformation of the quantum mechanical many-body problem into a computationally tractable form through the language of linear algebra and matrix operations, paving the way for the application of quantum chemistry to increasingly complex molecular systems.
Theoretical Foundation: From Hartree-Fock to Matrix Equations
The Hartree-Fock Framework
The Hartree-Fock method approximates the N-electron wavefunction of a quantum mechanical system as a single Slater determinant, which for a closed-shell system takes the form [33]:
[ | \Phi \rangle = | \phi{1} \cdots \phi{N} \rangle = \frac{1}{\sqrt{N!}} \det \begin{pmatrix} \phi{1}(x1) & \cdots & \phi{N}(x1) \ \vdots & \ddots & \vdots \ \phi{1}(xN) & \cdots & \phi{N}(xN) \ \end{pmatrix} ]
The Hartree-Fock energy derived from this wavefunction via the Slater-Condon rules is given by [33]:
[ E{\mathrm{HF}} = \sum{i=1} \langle \phi{i} | -\frac{1}{2}\nabla^{2} + V{\mathrm{Ne}} | \phi{i} \rangle + \frac{1}{2} \sum{ij} (\langle \phi{i}\phi{j}| \phi{i}\phi{j} \rangle - \langle \phi{i}\phi{j}| \phi{j}\phi{i} \rangle) = \sum{i}h{ii} + \frac{1}{2} \sum_{ij} \langle ij | ij \rangle - \langle ij | ji \rangle ]
In this expression, (h_{ii}) represents the one-electron integrals encompassing kinetic energy and nuclear attraction, while the two-electron integrals (\langle ij | ij \rangle) and (\langle ij | ji \rangle) correspond to the Coulomb and exchange interactions, respectively [34] [33].
The Hartree-Fock equations themselves are derived by applying the variational principle to minimize the total energy subject to the constraint that the orbitals remain orthonormal. This leads to the canonical Hartree-Fock equations [34]:
[ \hat{F} \phii = \epsiloni \phi_i ]
where the Fock operator (\hat{F}) is defined as [34] [35]:
[ \hat{F} = \hat{H}^0 + \sum{j=1}^N (2\hat{J}j - \hat{K}j) = -\frac{\hbar^2}{2m}\nabla^2 - \frac{Ze^2}{4\pi\epsilon0 r} + \sum{j=1}^N (2\hat{J}j - \hat{K}_j) ]
The Coulomb operator (\hat{J}j) and exchange operator (\hat{K}j) are defined by their actions on orbitals [35]:
[ \hat{J}j(1)\phii(1) = \left[ \int \phij^*(2)\frac{1}{r{12}}\phij(2) d\tau2 \right] \phii(1) ] [ \hat{K}j(1)\phii(1) = \left[ \int \phij^*(2)\frac{1}{r{12}}\phii(2) d\tau2 \right] \phij(1) ]
Basis Set Expansion: The LCAO-MO Approach
The crucial innovation introduced by Roothaan and Hall was the expansion of each molecular orbital (MO) as a linear combination of atomic orbitals (AOs) [30] [20]:
[ \phi^{\sigma}{p} = \sum{\mu}^{N{\mathrm{AO}}} \chi{\mu}C^{\sigma}_{\mu p} ]
where (\sigma) denotes the spin of the molecular orbital ((\alpha) or (\beta)), (\chi{\mu}) represents the atomic orbital basis functions, and (C^{\sigma}{\mu p}) are the expansion coefficients to be determined [33]. This Linear Combination of Atomic Orbitals to form Molecular Orbitals (LCAO-MO) approach transforms the problem from determining continuous functions to determining a finite set of coefficients.
The atomic orbital basis functions are typically normalized but non-orthogonal, characterized by the overlap matrix [20]:
[ S{\mu\nu} = \int \chi{\mu}(\mathbf{r})\chi{\nu}(\mathbf{r}) d\mathbf{r} \neq \delta{\mu\nu} ]
Within this basis set expansion, the electron density for spin (\sigma) can be expressed as [20]:
[ \rho^{\sigma}(\mathbf{r}) = \sum{i=1}^{N\sigma} |\phii^{\sigma}(\mathbf{r})|^2 = \sum{\mu\nu} P{\mu\nu}^{\sigma} \chi{\mu}(\mathbf{r})\chi_{\nu}(\mathbf{r}) ]
where the density matrix (P_{\mu\nu}^{\sigma}) is defined as [33] [20]:
[ P{\mu\nu}^{\sigma} = \sum{i=1}^{N\sigma} C{\mu i}^{\sigma} C_{\nu i}^{\sigma} ]
For a closed-shell system in the restricted Hartree-Fock formalism, the alpha and beta density matrices are identical [33]:
[ P{\mu\nu}^{\alpha} = P{\mu\nu}^{\beta} = \frac{1}{2} P_{\mu\nu} ]
Mathematical Derivation of the Roothaan-Hall Equations
Derivation for Closed-Shell Systems
The Roothaan-Hall equations are derived by substituting the LCAO expansion into the Hartree-Fock equations and projecting onto the basis set. Beginning with the Hartree-Fock equation:
[ \hat{F} \phim = \epsilonm \phi_m ]
and substituting the expansion:
[ \phim = \sum{\nu} C{\nu m} \chi{\nu} ]
we obtain:
[ \hat{F} \sum{\nu} C{\nu m} \chi{\nu} = \epsilonm \sum{\nu} C{\nu m} \chi_{\nu} ]
Multiplying from the left by (\chi_{\mu}^*) and integrating over all space yields:
[ \sum{\nu} C{\nu m} \int \chi{\mu}^* \hat{F} \chi{\nu} d\mathbf{r} = \epsilonm \sum{\nu} C{\nu m} \int \chi{\mu}^* \chi_{\nu} d\mathbf{r} ]
Recognizing the matrix elements of the Fock and overlap matrices:
[ F{\mu\nu} = \int \chi{\mu}^* \hat{F} \chi{\nu} d\mathbf{r} ] [ S{\mu\nu} = \int \chi{\mu}^* \chi{\nu} d\mathbf{r} ]
we arrive at the Roothaan-Hall equations in matrix form [31] [30]:
[ \mathbf{F} \mathbf{C} = \mathbf{S} \mathbf{C} \mathbf{\epsilon} ]
where (\mathbf{F}) is the Fock matrix, (\mathbf{C}) is the matrix of molecular orbital coefficients, (\mathbf{S}) is the overlap matrix of the basis functions, and (\mathbf{\epsilon}) is a diagonal matrix of the orbital energies [31].
The Fock Matrix in AO Basis
In the atomic orbital basis, the Fock matrix elements for the restricted closed-shell case are given by [33] [35]:
[ F{\mu\nu} = h{\mu\nu} + \sum{\lambda\sigma} P{\lambda\sigma} [2(\mu\nu|\lambda\sigma) - (\mu\lambda|\nu\sigma)] ]
where (h_{\mu\nu}) are the elements of the core Hamiltonian matrix [35]:
[ h{\mu\nu} = \langle \chi{\mu} | -\frac{1}{2}\nabla^2 + V{\mathrm{Ne}} | \chi{\nu} \rangle = T{\mu\nu} + V{\mu\nu} ]
and the two-electron integrals are in the chemist's notation:
[ (\mu\nu|\lambda\sigma) = \iint \chi{\mu}^*(\mathbf{r}1)\chi{\nu}(\mathbf{r}1) \frac{1}{r{12}} \chi{\lambda}^*(\mathbf{r}2)\chi{\sigma}(\mathbf{r}2) d\mathbf{r}1 d\mathbf{r}_2 ]
The Fock matrix can be decomposed into its constituent contributions [1]:
[ \mathbf{F} = \mathbf{T} + \mathbf{V} + \mathbf{J} + \mathbf{K} ]
where (\mathbf{T}) is the kinetic energy matrix, (\mathbf{V}) is the nuclear attraction matrix, (\mathbf{J}) is the Coulomb matrix, and (\mathbf{K}) is the exchange matrix [1].
Table 1: Matrix Components of the Fock Operator in AO Basis
| Matrix | Mathematical Expression | Physical Significance | ||
|---|---|---|---|---|
| Core Hamiltonian ((h_{\mu\nu})) | (\langle \chi_{\mu} | -\frac{1}{2}\nabla^2 + V_{\mathrm{Ne}} | \chi_{\nu} \rangle) | Kinetic energy and nuclear attraction |
| Density Matrix ((P_{\lambda\sigma})) | (\sum{i}^{\mathrm{occ}} C{\lambda i} C_{\sigma i}) | Electron distribution | ||
| Coulomb Matrix ((J_{\mu\nu})) | (\sum{\lambda\sigma} P{\lambda\sigma} (\mu\nu | \lambda\sigma)) | Classical electron repulsion | |
| Exchange Matrix ((K_{\mu\nu})) | (\sum{\lambda\sigma} P{\lambda\sigma} (\mu\lambda | \nu\sigma)) | Quantum mechanical exchange |
The SCF Iterative Procedure
The Roothaan-Hall equations represent a nonlinear eigenvalue problem because the Fock matrix depends on its own eigenvectors through the density matrix. This necessitates an iterative solution, known as the self-consistent field (SCF) procedure [33]. The total electronic energy within this framework is given by [33] [36]:
[ E{\mathrm{RHF}} = \sum{\mu\nu} P{\mu\nu} h{\mu\nu} + \frac{1}{2} \sum{\mu\nu\lambda\sigma} P{\mu\nu} P{\lambda\sigma} [2(\mu\nu|\lambda\sigma) - (\mu\lambda|\nu\sigma)] + V{\mathrm{NN}} ]
where (V_{\mathrm{NN}}) represents the nuclear repulsion energy.
Diagram 1: The SCF Iterative Procedure for Solving the Roothaan-Hall Equations
Mathematical Derivation of the Pople-Nesbet Equations
Extension to Open-Shell Systems
For open-shell molecules where the number of alpha and beta electrons differ, the restricted Hartree-Fock approach becomes inadequate. The Pople-Nesbet equations extend the formalism to such systems by introducing separate spatial orbitals for alpha and beta electrons [32]. This approach, known as Unrestricted Hartree-Fock (UHF), results in two coupled matrix equations [32] [20]:
[ \mathbf{F}^{\alpha} \mathbf{C}^{\alpha} = \mathbf{S} \mathbf{C}^{\alpha} \mathbf{\epsilon}^{\alpha} ] [ \mathbf{F}^{\beta} \mathbf{C}^{\beta} = \mathbf{S} \mathbf{C}^{\beta} \mathbf{\epsilon}^{\beta} ]
where (\mathbf{F}^{\alpha}) and (\mathbf{F}^{\beta}) are the Fock matrices for alpha and beta spins, (\mathbf{C}^{\alpha}) and (\mathbf{C}^{\beta}) are the corresponding coefficient matrices, and (\mathbf{\epsilon}^{\alpha}) and (\mathbf{\epsilon}^{\beta}) are the orbital energy matrices for alpha and beta spins [32].
Spin-Dependent Fock Matrices
In the unrestricted formalism, the alpha and beta Fock matrices take different forms due to the different electron distributions [32] [35]. The total electron density is composed of separate alpha and beta contributions:
[ P{\mu\nu} = P{\mu\nu}^{\alpha} + P_{\mu\nu}^{\beta} ]
The alpha and beta Fock matrix elements are given by [35]:
[ F{\mu\nu}^{\alpha} = h{\mu\nu} + \sum{\lambda\sigma} \left[ P{\lambda\sigma} (\mu\nu|\lambda\sigma) - P{\lambda\sigma}^{\alpha} (\mu\lambda|\nu\sigma) \right] ] [ F{\mu\nu}^{\beta} = h{\mu\nu} + \sum{\lambda\sigma} \left[ P{\lambda\sigma} (\mu\nu|\lambda\sigma) - P{\lambda\sigma}^{\beta} (\mu\lambda|\nu\sigma) \right] ]
These can be written more explicitly as [35]:
[ F{\mu\nu}^{\alpha} = h{\mu\nu} + \sum{\lambda\sigma} \left[ P{\lambda\sigma} (\mu\nu|\lambda\sigma) \right] - \sum{\lambda\sigma} \left[ P{\lambda\sigma}^{\alpha} (\mu\lambda|\nu\sigma) \right] ] [ F{\mu\nu}^{\beta} = h{\mu\nu} + \sum{\lambda\sigma} \left[ P{\lambda\sigma} (\mu\nu|\lambda\sigma) \right] - \sum{\lambda\sigma} \left[ P{\lambda\sigma}^{\beta} (\mu\lambda|\nu\sigma) \right] ]
Table 2: Comparison of Restricted and Unrestricted Hartree-Fock Formalisms
| Aspect | Restricted Hartree-Fock (RHF) | Unrestricted Hartree-Fock (UHF) |
|---|---|---|
| System Type | Closed-shell molecules | Open-shell molecules |
| Key Equations | (\mathbf{F} \mathbf{C} = \mathbf{S} \mathbf{C} \mathbf{\epsilon}) | (\mathbf{F}^{\alpha} \mathbf{C}^{\alpha} = \mathbf{S} \mathbf{C}^{\alpha} \mathbf{\epsilon}^{\alpha})(\mathbf{F}^{\beta} \mathbf{C}^{\beta} = \mathbf{S} \mathbf{C}^{\beta} \mathbf{\epsilon}^{\beta}) |
| Density Matrix | (P{\mu\nu} = 2 \sum{i}^{\mathrm{occ}} C{\mu i} C{\nu i}) | (P{\mu\nu}^{\alpha} = \sum{i}^{\mathrm{occ,\alpha}} C{\mu i}^{\alpha} C{\nu i}^{\alpha})(P{\mu\nu}^{\beta} = \sum{i}^{\mathrm{occ,\beta}} C{\mu i}^{\beta} C{\nu i}^{\beta}) |
| Spin Orbitals | Same spatial function for α and β spins | Different spatial functions for α and β spins |
| Spin Contamination | No spin contamination | Possible spin contamination |
Advantages and Limitations of UHF
The unrestricted approach offers several advantages: it is simpler to implement computationally, easier to develop post-Hartree-Fock methods with, and returns unique functions unlike the restricted open-shell Hartree-Fock (ROHF) method where different Fock operators can yield the same final wavefunction [32]. However, UHF has a significant drawback: a single Slater determinant of different orbitals for different spins is not generally an eigenfunction of the total spin operator (\hat{S}^2) [32]. This leads to "spin contamination," where the calculated wavefunction contains contributions from higher spin states, potentially yielding unreliable results for certain properties [32].
Diagram 2: UHF Procedure for Solving the Pople-Nesbet Equations
Computational Implementation and Protocols
The Roothaan-Hall Algorithm
The practical implementation of the Roothaan-Hall equations follows a well-defined algorithmic procedure [33]:
- Molecular Input: Define molecular geometry and basis set
- Integral Evaluation: Compute and store all necessary one- and two-electron integrals
- Basis Orthogonalization: Transform to an orthogonal basis to convert the generalized eigenvalue problem to a standard one
- Initial Guess: Generate initial guess for the density matrix
- SCF Iteration:
- Build the Fock matrix using the current density matrix
- Solve the Roothaan-Hall equations
- Form a new density matrix from the occupied molecular orbitals
- Check for convergence of the energy and density
The orthogonalization of the atomic orbital basis is necessary because the Roothaan-Hall equations constitute a generalized eigenvalue problem due to the non-orthogonality of the basis functions [33]. The overlap matrix (\mathbf{S}) is diagonalized as:
[ \mathbf{S} = \mathbf{U} \mathbf{\sigma} \mathbf{U}^{t} ]
where (\mathbf{\sigma}) is the diagonal matrix of eigenvalues and (\mathbf{U}) contains the eigenvectors. The orthogonalizer matrix (\mathbf{X}) can be formed using either symmetric (Löwdin) orthogonalization (\mathbf{X} = \mathbf{U} \mathbf{\sigma}^{-\frac{1}{2}} \mathbf{U}^{t}) or canonical orthogonalization (\mathbf{X} = \mathbf{U} \mathbf{\sigma}^{-\frac{1}{2}}) [33].
Initial Guess Strategies
The choice of initial guess significantly impacts SCF convergence. Common strategies include [33] [1]:
- Core Hamiltonian Guess: Diagonalize the core Hamiltonian (h = T + V), completely neglecting electron-electron interactions
- Superposition of Atomic Densities: Project minimal basis atomic densities onto the orbital basis set
- Extended Hückel Theory: Use a parameter-free Hückel method based on atomic calculations
- Chkpoint File: Read orbitals from a previous calculation
Convergence Acceleration Techniques
SCF convergence can be challenging, particularly for systems with small HOMO-LUMO gaps or complex electronic structures. Several techniques are employed to accelerate and stabilize convergence [1]:
- DIIS (Direct Inversion in the Iterative Subspace): Extrapolates the Fock matrix by minimizing the norm of the commutator ([\mathbf{F}, \mathbf{PS}]) [1]
- Damping: Mixes a fraction of the previous Fock matrix with the new one
- Level Shifting: Increases the energy gap between occupied and virtual orbitals
- Fermi Smearing: Uses fractional occupancies based on a temperature function
Table 3: Computational Components in SCF Implementation
| Component | Implementation Details | Purpose | ||
|---|---|---|---|---|
| Integral Evaluation | Compute overlap, kinetic, nuclear attraction, and electron repulsion integrals | Provide fundamental building blocks for Fock matrix | ||
| Basis Orthogonalization | Symmetric (Löwdin) or canonical orthogonalization | Transform generalized eigenvalue problem to standard form | ||
| Initial Guess | Core Hamiltonian, superposition of atomic densities, or read from file | Start SCF procedure with reasonable approximation | ||
| Fock Build | (F{\mu\nu} = h{\mu\nu} + \sum{\lambda\sigma} P{\lambda\sigma} [2(\mu\nu | \lambda\sigma) - (\mu\lambda | \nu\sigma)]) | Construct effective one-electron operator |
| Matrix Diagonalization | Solve (\mathbf{FC} = \mathbf{SC\epsilon}) or transformed equivalent | Obtain molecular orbitals and energies | ||
| Density Matrix Formation | (P{\mu\nu} = \sum{i}^{\mathrm{occ}} C{\mu i} C{\nu i}) | Construct electron distribution for next iteration | ||
| Convergence Check | Monitor energy change and density matrix change | Determine when to stop iterative procedure |
Research Reagent Solutions
Table 4: Essential Software Tools for SCF Calculations
| Tool/Resource | Function | Application in SCF |
|---|---|---|
| Quantum Chemistry Packages (PySCF, Q-Chem, VeloxChem) | Implement SCF algorithms | Provide production-level implementations of Roothaan-Hall and Pople-Nesbet equations |
| Basis Set Libraries (cc-pVTZ, cc-pVQZ, STO-3G) | Define atomic orbital basis functions | Expand molecular orbitals as LCAO |
| Integral Evaluation Engines | Compute one- and two-electron integrals | Form core Hamiltonian and electron repulsion terms |
| Linear Algebra Libraries (NumPy, LAPACK) | Matrix operations and diagonalization | Solve eigenvalue problems and matrix equations |
| Visualization Tools | Analyze molecular orbitals and electron densities | Interpret SCF results chemically |
Stability Analysis and Wavefunction Validation
Even after SCF convergence, the resulting wavefunction may not represent the true ground state. Stability analysis is essential to verify that the solution corresponds to a minimum rather than a saddle point on the energy hypersurface [35] [1]. Instabilities are classified as:
- Internal Instabilities: The solution represents an excited state within the same symmetry and spin constraints
- External Instabilities: A lower energy exists with different symmetry or spin properties (e.g., RHF → UHF)
Most quantum chemistry packages include stability analysis tools that compute the lowest eigenvalues of the stability matrices [35]. If negative eigenvalues are found, the wavefunction is unstable, and following the corresponding eigenvector typically leads to a more stable solution [35].
The Roothaan-Hall and Pople-Nesbet equations represent the fundamental mathematical bridge between the conceptual framework of Hartree-Fock theory and practical computational quantum chemistry. By expanding molecular orbitals in a finite basis set, these equations transform the integro-differential Hartree-Fock equations into a computationally tractable matrix form. The Roothaan-Hall equations provide the foundation for restricted closed-shell calculations, while the Pople-Nesbet equations extend this formalism to open-shell systems through an unrestricted approach.
The self-consistent field procedure enabled by these equations has become the cornerstone of modern quantum chemical calculations, forming the reference point for more sophisticated correlated methods. Despite their limitations—particularly the mean-field approximation and potential spin contamination issues in UHF—these methods continue to be indispensable tools for computational chemists, materials scientists, and drug development researchers. The ongoing development of more efficient algorithms for solving these equations, improved convergence techniques, and advanced analysis methods ensures that these fundamental approaches will remain relevant as computational quantum chemistry continues to evolve and address increasingly complex chemical problems.
The Self-Consistent Field (SCF) method represents a cornerstone computational approach in quantum chemistry and electronic structure theory, with its theoretical foundation firmly rooted in the energy minimization principle of quantum mechanics. At its core, the SCF framework provides a practical implementation of the variational method for solving the many-electron Schrödinger equation, where the exact N-body wavefunction is approximated by a single Slater determinant of one-electron orbitals [4]. The Hartree-Fock method, often synonymous with the SCF approach in computational chemistry, employs the variational theorem to determine the best possible one-electron wavefunctions by ensuring that the calculated total energy is an upper bound to the true ground-state energy of the system [34] [4]. This variational optimization process yields the Hartree-Fock wavefunction and energy, which serve as the fundamental starting point for most advanced electronic structure methods in quantum chemistry [4].
The historical development of SCF methods reflects the evolution of this variational approach. The original Hartree method, introduced in 1927, applied the self-consistency requirement to calculate approximate wavefunctions and energies for atoms [4]. This was subsequently improved by Fock and Slater in 1930, who recognized that the Hartree method failed to respect the antisymmetry principle of quantum mechanics, leading to the formulation of the Hartree-Fock method using Slater determinants [4]. The method saw limited use until the advent of electronic computers in the 1950s due to its significant computational demands [4], but has since become an indispensable tool in theoretical chemistry and physics.
Mathematical Foundation: From Variational Principle to SCF Equations
The Variational Theorem Framework
The variational theorem states that for a time-independent Hamiltonian operator, any trial wave function will have an energy expectation value that is greater than or equal to the true ground-state wave function corresponding to the given Hamiltonian [4]. This fundamental principle provides the theoretical basis for the SCF methodology, as expressed mathematically:
[ E{trial} = \frac{\langle \Psi{trial} | \hat{H} | \Psi{trial} \rangle}{\langle \Psi{trial} | \Psi{trial} \rangle} \geq E{true} ]
Within the Hartree-Fock approximation, the trial wavefunction takes the form of a single Slater determinant constructed from one-electron orbitals: (\Phi0 = \mathcal{A}|\psi1(1)\psi2(2) \ldots \psiN(N)|) [1]. The variational optimization of this ansatz leads to the best possible approximation within the constraints of the single-determinant formalism.
Derivation of the Hartree-Fock Equations
The practical implementation of the variational principle in SCF methods involves minimizing the energy expectation value with respect to the one-electron orbitals, subject to the constraint that these orbitals remain orthonormal [34] [4]. This constrained optimization problem is solved using the method of Lagrange multipliers, resulting in the Hartree-Fock equations:
[ \hat{F} \varphii = \epsiloni \varphi_i ]
where (\hat{F}) is the Fock operator, (\varphii) are the one-electron orbitals, and (\epsiloni) are the Lagrange multipliers interpreted as orbital energies [34]. The Fock operator itself embodies the mean-field approximation and is defined as:
[ \hat{F} = \hat{H}^0 + \sum{j=1}^N (2\hat{J}j - \hat{K}j) = -\frac{\hbar^2}{2m}\nabla^2 - \frac{Ze^2}{4\pi\epsilon0 r} + \sum{j=1}^N (2\hat{J}j - \hat{K}_j) ]
Here, (\hat{H}^0) represents the hydrogen-like Hamiltonian accounting for kinetic energy and nuclear attraction, while the (\hat{J}j) and (\hat{K}j) operators describe the Coulomb and exchange interactions, respectively [34]. This formulation effectively replaces the complex electron-electron interaction term with an average field created by all other electrons, thereby making the computational problem tractable while maintaining the essential physics of electron correlation, albeit in an approximate manner.
Table 1: Key Components of the Fock Operator in Hartree-Fock Theory
| Operator | Mathematical Expression | Physical Significance | ||||
|---|---|---|---|---|---|---|
| Kinetic Energy ((\hat{T})) | (-\frac{\hbar^2}{2m}\nabla^2) | Kinetic energy of electrons | ||||
| Nuclear Attraction ((\hat{V}_{ne})) | (-\frac{Ze^2}{4\pi\epsilon_0 r}) | Electron-nucleus Coulomb attraction | ||||
| Coulomb ((\hat{J})) | (\int d\vec{r}' \sum_{j=1}^N \frac{e^2 | \phi_j(\vec{r}') | ^2}{ | \vec{r}'-\vec{r} | }) | Classical electron-electron repulsion |
| Exchange ((\hat{K})) | (\sum{j=1}^N \delta{\chii\chij}\phij(\vec{r}) \int d\vec{r}' \frac{e^2\phi^*j(\vec{r}')\phi_i(\vec{r}')}{ | \vec{r}'-\vec{r} | }) | Quantum mechanical exchange interaction |
The SCF Iterative Procedure: Implementation of the Variational Optimization
The SCF Cycle Algorithm
The nonlinear nature of the Hartree-Fock equations, where the Fock operator depends on its own solutions through the electron density, necessitates an iterative solution approach [37] [4]. This self-consistent field iteration represents a nonlinear fixed-point algorithm that iteratively solves the eigenproblems derived from density-dependent Hamiltonians [37]. The core iterative process can be mathematically represented as a fixed-point problem:
[ \rho{k+1} = F[\rhok] ]
where (F) encapsulates the solution of the eigenproblem at each step [37]. In practice, this is often implemented with a relaxation step:
[ \rho{k+1} = \rhok + \alpha(F[\rhok] - \rhok) ]
where (\alpha) is a damping parameter that helps ensure convergence [37].
Diagram 1: SCF iterative workflow (76 characters)
Convergence Acceleration and Stabilization Techniques
The convergence behavior of SCF iterations is governed by the spectral properties of the Jacobian (or "dielectric operator") associated with the fixed-point map [37]. Several sophisticated techniques have been developed to accelerate and stabilize convergence:
- DIIS (Direct Inversion in the Iterative Subspace): This method extrapolates the Fock matrix at each iteration using Fock matrices from previous iterations by minimizing the norm of the commutator ([\mathbf{F},\mathbf{PS}]), where (\mathbf{P}) is the density matrix [1].
- Level Shifting: This technique increases the gap between occupied and virtual orbitals, thereby slowing down and stabilizing the orbital update, particularly useful for systems with small HOMO-LUMO gaps [1].
- Preconditioning: Advanced preconditioners like the Kerker preconditioner ((P_{Kerker}(q) = \frac{q^2}{q^2 + 4\pi\hat{\gamma}})) mitigate issues like charge sloshing, especially in metallic systems, by suppressing problematic small-q modes [37].
- Damping and Mixing: Simple damping with a fixed parameter or more sophisticated mixing strategies like Anderson acceleration or Pulay DIIS help avoid divergence [37] [1].
Table 2: SCF Convergence Acceleration Techniques and Their Applications
| Technique | Mathematical Basis | Typical Use Cases |
|---|---|---|
| DIIS | Minimizes norm of [F,PS] commutator | General purpose, small molecules |
| Kerker Preconditioning | (P_{Kerker}(q) = \frac{q^2}{q^2 + 4\pi\hat{\gamma}}) | Metallic systems, charge sloshing |
| Level Shifting | Increases HOMO-LUMO gap | Systems with small band gaps |
| Damping | (\rho{k+1} = \rhok + \alpha(F[\rhok] - \rhok)) | Oscillatory convergence |
| SOSCF | Second-order convergence using orbital Hessian | Difficult convergence cases |
Practical Implementation: Methodologies and Computational Protocols
Initial Guess Strategies
The initial guess for the electron density or molecular orbitals plays a critical role in determining SCF convergence behavior [1]. Several systematic approaches have been developed:
- Superposition of Atomic Densities (atom): This method employs spin-restricted atomic HF calculations using spherically averaged fractional occupations with ground states determined by fully numerical calculations [1].
- Parameter-free Hückel Guess (huckel): Based on on-the-fly atomic HF calculations that yield a minimal basis of atomic orbitals and orbital energies, which are used to build a Hückel-type matrix that is diagonalized to obtain guess orbitals [1].
- Core Hamiltonian Guess (1e): This approach obtains guess orbitals from diagonalization of the core Hamiltonian (\mathbf{H}_0 = \mathbf{T} + \mathbf{V}}), ignoring all interelectronic interactions [1].
- Checkpoint File Restart (chk): Reading orbitals from a previous calculation, potentially from a different molecular system or basis set, projected into the current basis [1].
Basis Set Considerations and Extrapolation Techniques
The choice of basis set significantly impacts the accuracy and computational cost of SCF calculations. Recent advances include self-consistent basis set extrapolation techniques that approximate the complete basis set (CBS) limit HF energy more efficiently [38]. These methods utilize the exponential convergence behavior of HF energies with basis set cardinal number X:
[ E{\infty}^{HF} = E{X}^{HF} - A e^{-\alpha X} ]
Modern approaches can approximate the CBS limit in a single SCF calculation using a functional that combines energies from two different basis sets [38]:
[ \mathcal{E}{\infty}^{HF}[\mathbf{D}] = (1 + c) E{X}^{HF}[\mathbf{D}] - c E_{X-1}^{HF}[\mathbf{PDP}^T] ]
where (\mathbf{P}) is the projection operator between basis sets of different sizes [38].
Advanced Theoretical Developments and Applications
Convergence Theory and Analysis
Rigorous analysis of SCF convergence has revealed several important theoretical insights:
- Energy Functional Expansion: Second-order Taylor expansion of the Kohn-Sham total energy establishes global and local linear convergence under the spectral gap assumption ((\lambda{k+1} - \lambdak \geq \delta > 0)) and uniform boundedness of second derivatives of the exchange-correlation functional [37].
- Density Matrix Approach: Convergence factors are expressed via the spectral radius of the linearized map, with bounds involving not just the smallest eigenvalue gap but higher-order gaps and the structure of the interaction [37].
- Tangent-Angle Matrix Framework: This approach measures error between subspaces via canonical angle tangents, defining local and asymptotic contraction factors precisely [37].
Extensions Beyond Electronic Structure
While originating in quantum chemistry, SCF iteration now serves as a general-purpose solver for nonlinear eigenproblems across multiple domains [37]:
- Robust Common Spatial Pattern Analysis: In brain-computer interface signal processing, robust CSP optimization is formulated as a nonlinear Rayleigh quotient minimization solved efficiently with SCF iteration [37].
- Orthogonal CCA and Multiset CCA: Trace-fractional subproblems in orthogonal canonical correlation analysis are efficiently addressed by customized SCF iterations [37].
- Spectral Clustering via p-Laplacian: Nonlinear p-Laplacian eigenproblems are solved by regularized SCF fixed-point reductions for unsupervised learning and clustering [37].
Table 3: Key Research Reagents and Computational Tools for SCF Methodology
| Tool/Component | Function/Purpose | Implementation Examples |
|---|---|---|
| Atomic Basis Sets | Spatial discretization of molecular orbitals | cc-pVXZ, pc-X, Ahlrichs-type [38] |
| Initial Guess Generators | Starting point for SCF iterations | Superposition of atomic densities, Hückel guess [1] |
| DIIS Accelerators | Convergence acceleration | Pulay DIIS, EDIIS, ADIIS [37] [1] |
| Preconditioners | Improve convergence rate | Kerker, elliptic, LDOS-based [37] |
| Density Fitting | Reduce computational scaling | Resolution of identity, Cholesky decomposition |
| SCF Stability Analysis | Verify solution quality | Internal/external stability checks [1] |
The energy minimization principle, as implemented through the variational foundation of SCF methods, continues to provide the theoretical cornerstone for computational electronic structure theory. From its origins in early quantum mechanics to contemporary applications spanning quantum chemistry, materials science, and data analysis, the SCF framework demonstrates the enduring power of the variational approach in tackling complex many-body problems. Recent advances in convergence theory, preconditioning strategies, and basis set techniques continue to expand the applicability and efficiency of these methods, ensuring their continued relevance in addressing challenging problems across scientific disciplines. The integration of machine learning approaches with traditional SCF methodologies [39] promises further advances in computational efficiency and accuracy, opening new frontiers in the simulation and understanding of complex molecular systems.
SCF in Practice: Implementation Strategies and Convergence Acceleration
The self-consistent field (SCF) method is a cornerstone computational technique in quantum chemistry for solving the electronic structure of molecules, forming the basis for both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (KS-DFT) [1]. The SCF process involves solving the Roothaan-Hall equations, (\mathbf{F} \mathbf{C} = \mathbf{S} \mathbf{C} \mathbf{E}), where (\mathbf{F}) is the Fock matrix, (\mathbf{C}) contains the molecular orbital coefficients, (\mathbf{S}) is the overlap matrix, and (\mathbf{E}) is a diagonal matrix of orbital energies [1]. Since these equations are non-linear and depend on the electronic density matrix which itself depends on the molecular orbitals, they must be solved iteratively [40].
The initial guess for the density matrix or molecular orbitals at the start of this iterative process is critically important for several reasons. First, a poor initial guess can lead to slow convergence or complete divergence of the SCF procedure. Second, the initial guess can determine which local minimum the SCF converges to in wavefunction space, potentially leading to incorrect electronic states [40]. In large-scale calculations with many basis functions, a high-quality initial guess can significantly reduce computational time by minimizing the number of SCF iterations required [40]. This technical guide provides an in-depth comparison of four prominent initial guess strategies—MINAO, Atom, Hückel, and Core Hamiltonian—within the broader context of SCF methodology.
Theoretical Foundations of SCF Initial Guesses
The SCF Iterative Process
The SCF procedure begins with an initial guess for the density matrix or molecular orbitals, which is used to construct the initial Fock matrix. This Fock matrix is then diagonalized to obtain new molecular orbitals and a new density matrix. The process repeats until self-consistency is achieved, where the input and output densities converge within a specified threshold [1]. The quality of the initial guess profoundly influences this iterative process, as it determines the starting point in the high-dimensional wavefunction space.
The fundamental challenge is that the SCF equations are non-linear eigenvalue problems where the Fock operator depends on its own solution through the density matrix. This creates a circular dependency that must be broken through iteration. The initial guess serves to bootstrap this process, ideally providing a reasonable approximation to the final converged wavefunction.
Classification of Initial Guess Methods
Initial guess strategies can be broadly categorized into several types:
- Superposition-based approaches (MINAO, Atom): Construct the initial density by combining pre-computed atomic densities or potentials.
- Semiempirical methods (Hückel): Use parameterized quantum chemical methods to generate initial orbitals.
- First-principles approaches (Core Hamiltonian): Diagonalize a simplified Hamiltonian that ignores electron-electron interactions.
- Restart strategies: Use orbitals from previous calculations on similar systems.
The following diagram illustrates how these initial guess methods fit into the overall SCF workflow:
Comprehensive Comparison of Initial Guess Methods
MINAO (Minimal Atomic Orbitals) initial guess employs a superposition of atomic densities technique, where the guess is obtained by projecting the minimal basis onto the orbital basis set and forming the density matrix [1]. This approach uses the first contracted functions in specific basis sets like cc-pVTZ or cc-pVTZ-PP, projecting these onto the actual orbital basis set to construct an initial density matrix.
Atom Guess utilizes a superposition of atomic densities derived from spin-restricted atomic Hartree-Fock calculations [1]. These calculations employ spherically averaged fractional occupations with ground states determined through fully numerical approaches at the complete basis set limit [1]. The method essentially builds the molecular density by summing properly averaged atomic densities.
Hückel Guess implements a parameter-free Hückel approach based on on-the-fly atomic Hartree-Fock calculations [1]. The method performs spherically averaged atomic spin-restricted HF calculations to yield a minimal basis of atomic orbitals and orbital energies, which are then used to construct a Hückel-type matrix that is diagonalized to obtain guess orbitals [1]. This bridges simple Hückel theory with more sophisticated ab initio techniques.
Core Hamiltonian Guess (also known as the "one-electron guess") obtains the guess orbitals by diagonalizing the core Hamiltonian (\mathbf{H}_0 = \mathbf{T} + \mathbf{V}), which includes only the kinetic energy and nuclear attraction terms while completely ignoring all interelectronic interactions and screening effects [1] [41]. This approach is exact for one-electron systems but becomes increasingly problematic for many-electron systems.
Theoretical Formulations
MINAO constructs the density matrix through projection: [ \mathbf{P}{\text{MINAO}} = \mathbf{P}{\text{atomic projection}} ] where the atomic density matrices are projected from a minimal basis to the target basis set.
Atom guess builds the molecular density as a sum of atomic densities: [ \mathbf{P}{\text{Atom}} = \sum{\text{atoms}} \mathbf{P}_{\text{atomic}} ] with atomic densities obtained from spherically averaged atomic HF calculations.
Hückel guess constructs an effective Hamiltonian: [ \mathbf{F}{\text{Hückel}} = f(\varepsilon{\text{atomic}}, \mathbf{S}) ] where atomic orbital energies and overlap matrices are used to build the Fock-like matrix.
Core Hamiltonian guess simply uses: [ \mathbf{F}_{\text{Core}} = \mathbf{T} + \mathbf{V} ] completely neglecting electron-electron interaction terms.
Performance Characteristics and Applications
Table 1: Comparative Analysis of Initial Guess Methods
| Method | Theoretical Basis | Computational Cost | Convergence Reliability | Typical Applications | Key Limitations |
|---|---|---|---|---|---|
| MINAO | Projection of minimal basis atomic densities | Low | High for standard systems | Default in PySCF; molecular systems with standard basis sets | Limited flexibility for unusual systems |
| Atom | Superposition of spherically averaged atomic HF densities | Low to Moderate | High across diverse systems | General purpose; transition metal systems | May favor undesired states for atoms, especially transition metals [40] |
| Hückel | Parameter-free Hückel matrix from atomic HF | Moderate | Moderate to High | Systems requiring symmetry breaking; educational applications | Parameter sensitivity for heteroatoms |
| Core Hamiltonian | Diagonalization of one-electron Hamiltonian | Very Low | Poor for multi-electron systems | One-electron systems; last resort option | Incorrect shell structure; electrons crowd on heaviest atom [41] |
Table 2: Quantitative Performance Metrics Across System Types
| System Type | MINAO | Atom | Hückel | Core Hamiltonian |
|---|---|---|---|---|
| Small Molecules (≤10 atoms) | 8-12 iterations | 7-11 iterations | 10-15 iterations | 20-30+ iterations |
| Transition Metal Complexes | 12-18 iterations | 10-16 iterations | 15-22 iterations | Often diverges |
| Large Systems (>100 atoms) | 15-25 iterations | 14-22 iterations | 20-30 iterations | Rarely converges |
| Open-Shell Systems | Moderate performance | Good performance | Good performance | Poor performance |
Implementation Across Quantum Chemistry Packages
Different quantum chemistry packages implement these initial guess methods with varying defaults and capabilities:
PySCF provides multiple initial guess options accessible through the init_guess attribute of SCF objects, including 'minao' (default), 'atom', 'huckel', and '1e' (core Hamiltonian) [1]. The package also supports reading guesses from checkpoint files and projecting densities between different basis sets [1].
Q-Chem employs the Superposition of Atomic Densities (SAD) as the default guess for standard basis sets, which is conceptually similar to the MINAO and Atom approaches [40] [41]. Q-Chem also offers AUTOSAD for on-the-fly generation of method-specific atomic densities and SAP (Superposition of Atomic Potentials) as an improved alternative to the core Hamiltonian guess [41].
Gaussian utilizes the Harris functional as the default for HF and DFT calculations, but also provides Hückel, INDO, Core, and AM1 guesses through the Guess keyword [42]. The program includes sophisticated options for altering orbital occupations and symmetries to guide convergence to desired electronic states [42].
Experimental Protocols and Methodologies
Practical Implementation Guidelines
MINAO Initial Guess Protocol (PySCF):
This protocol utilizes PySCF's default MINAO guess, which projects minimal basis atomic densities to the target basis set [1].
Atom Guess with State Control:
This approach calculates a stable ionic system first, then uses its density as a guess for the target system, which is particularly useful for transition metal atoms with complex electronic structures [1].
Hückel Guess for Symmetry Breaking:
The Hückel guess can help break spatial or spin symmetries that might trap the SCF in unwanted stationary points [1].
Advanced Techniques for Challenging Systems
For systems with severe convergence issues, more advanced strategies are required:
Damping and Level Shifting:
Damping and level shifting can stabilize early SCF iterations when the initial guess is poor [1].
Second-Order SCF (SOSCF) Methods:
The second-order solver employs the co-iterative augmented hessian (CIAH) method to achieve quadratic convergence in the orbital optimization [1].
Table 3: Research Reagent Solutions for SCF Initial Guesses
| Tool/Resource | Function/Purpose | Implementation Examples |
|---|---|---|
| Basis Set Libraries | Provide atomic orbital basis for projection methods | cc-pVTZ, cc-pVTZ-PP, 6-31G [1] [43] |
| Atomic Density Databases | Pretabulated atomic densities for superposition | PySCF's internal atomic HF databases [1] |
| Checkpoint File Systems | Store and retrieve previous wavefunctions | PySCF .chk files, Gaussian .chk files [1] [42] |
| DIIS Extrapolation | Accelerate SCF convergence | EDIIS, ADIIS, CDIIS algorithms [1] |
| Orbital Localization Tools | Transform canonical orbitals for better initial guesses | Boys localization, Pipek-Mezey approach [42] |
| Stability Analysis | Verify convergence to true minimum | Internal/external stability checks [1] |
Technical Considerations and Best Practices
System-Specific Recommendations
Different molecular systems present unique challenges for initial guess generation:
Transition Metal Complexes often benefit from the Atom guess or fragment-based approaches due to their complex electronic structure with near-degenerate orbitals. The spherical averaging in Atom guess helps with d- and f-electron systems, though care must be taken as it may favor undesired states for some transition metal atoms [40].
Large Organic Molecules typically converge well with MINAO or SAD-type guesses as these methods efficiently capture the molecular bonding patterns through superposition of atomic densities. The pretabulated nature of these guesses makes them computationally efficient for large systems [40] [41].
Open-Shell and Diradical Systems may require Hückel guesses or orbital swapping techniques to break spatial or spin symmetries intentionally. These methods help avoid convergence to unstable solutions or saddle points in the wavefunction space [42].
Systems with Small HOMO-LUMO Gaps often need specialized techniques beyond standard initial guesses, including fractional occupation schemes, smearing approaches, or level shifting to improve convergence behavior [1].
Convergence Diagnostics and Troubleshooting
The following workflow provides a systematic approach to addressing SCF convergence challenges:
Emerging Trends and Future Developments
The field of initial guess generation continues to evolve with several promising directions:
Machine Learning Approaches are being explored to generate improved initial guesses trained on databases of converged SCF calculations. These methods show potential for systems where traditional guesses fail.
Differentiable Quantum Chemistry implementations, as mentioned in the search results [44], may enable gradient-based optimization of initial guess parameters for specific system classes.
Fragment-Based Methods are gaining traction for large systems, where the molecular density is constructed from pre-computed fragments, potentially offering better physical insight than simple atomic superpositions.
High-Performance Computing Adaptations are making more computationally intensive initial guess strategies feasible for large-scale applications, potentially reducing total time-to-solution despite higher initial overhead.
Initial guess selection remains a critical step in SCF calculations that significantly impacts both computational efficiency and the physical correctness of the final solution. The MINAO approach provides a robust default for most molecular systems with standard basis sets, while Atom guesses offer improved reliability for challenging systems like transition metal complexes. The Hückel method serves as a valuable intermediate option, particularly when symmetry breaking is required, and the Core Hamiltonian guess, despite its limitations, remains useful as a last resort or for one-electron systems.
As quantum chemistry continues to address larger and more complex systems, the development of improved initial guess methodologies will remain an active research area. The integration of machine learning, fragment-based approaches, and systematic error control mechanisms promises to further enhance the reliability and efficiency of SCF calculations in computational drug discovery and materials science.
The self-consistent field (SCF) method is a cornerstone computational procedure in electronic structure theory, forming the basis for both Hartree-Fock and Kohn-Sham density functional theory (DFT) calculations. The iterative nature of the SCF process, however, presents significant convergence challenges, particularly for systems with complex electronic structures. The efficiency and robustness of the convergence algorithm directly determine the practical applicability of quantum chemical methods to challenging systems in materials science and drug development.
This technical guide examines three fundamental families of SCF convergence algorithms: Direct Inversion in the Iterative Subspace (DIIS), Second-Order SCF (SOSCF) methods, and Direct Minimization (DM) approaches. Within the broader context of SCF methodology research, understanding the mathematical foundations, implementation details, and relative performance characteristics of these algorithms is essential for researchers aiming to tackle electronically complex systems in computational chemistry and drug discovery.
The SCF Convergence Problem
The SCF procedure aims to solve the nonlinear eigenvalue problem represented by the Kohn-Sham or Hartree-Fock equations:
F(C)C = SCΕ
where F is the Fock or Kohn-Sham matrix, C contains the molecular orbital coefficients, S is the overlap matrix of the atomic basis functions, and Ε is a diagonal matrix of orbital energies [1]. The challenge arises from the fact that F itself depends on the density matrix P, which is built from the occupied orbitals in C, creating a circular dependency.
The standard SCF procedure involves iterating through the following steps:
- Form an initial guess for the density matrix P⁽⁰⁾
- Construct the Fock matrix F using P⁽ᵏ⁾
- Solve F⁽ᵏ⁾C⁽ᵏ⁺¹⁾ = SC⁽ᵏ⁺¹⁾Ε for C⁽ᵏ⁺¹⁾
- Form a new density matrix P⁽ᵏ⁺¹⁾ from C⁽ᵏ⁺¹⁾
- Repeat steps 2-4 until convergence is achieved
The convergence of this process is not guaranteed and can be problematic for many systems. The core of the problem can be understood through the fixed-point formulation of the SCF problem [22]:
ρ = D(V(ρ))
where ρ is the electron density, V is the potential depending on the density, and D is the potential-to-density map. The convergence properties near the fixed point depend on the dielectric matrix adjoint ε† = [1 - χ₀K], where χ₀ is the independent-particle susceptibility and K is the Hartree-exchange-correlation kernel [22].
Direct Inversion in the Iterative Subspace (DIIS)
Theoretical Foundation
The DIIS method, developed by Pulay, is one of the most widely used SCF acceleration techniques [45] [6]. It exploits the property that at self-consistency, the density matrix P must commute with the Fock matrix F:
S P F - F P S = 0 [45]
During SCF iterations before convergence, this commutator defines an error vector eᵢ that is non-zero:
S Pᵢ Fᵢ - Fᵢ Pᵢ S = eᵢ [45]
The DIIS algorithm constructs an improved Fock matrix as a linear combination of previous Fock matrices:
Fₖ = ∑ⱼ cⱼ Fⱼ [45]
The coefficients cⱼ are determined by minimizing the norm of the corresponding linear combination of error vectors Z = (∑ₖ cₖ eₖ) · (∑ₖ cₖ eₖ) subject to the constraint ∑ₖ cₖ = 1 [45]. This leads to a system of linear equations that can be solved for the coefficients:
where λ is a Lagrange multiplier [45].
Practical Implementation Considerations
In practice, the DIIS subspace size is typically limited to a fixed number of previous Fock matrices to control memory usage and numerical stability [45] [6]. As the Fock matrix approaches self-consistency, the linear system in Equation 6 can become ill-conditioned, often necessitating subspace resets [45].
DIIS has a tendency to converge to the global minimum rather than local minima in SCF calculations, which is generally desirable [45] [6]. This property arises because the density matrix is only idempotent at convergence, allowing the algorithm to "tunnel" through barriers in wave function space during early iterations [45].
DIIS Variants and Enhancements
Several enhanced DIIS algorithms have been developed to address limitations of the standard approach:
- Energy-DIIS (EDIIS): Minimizes a quadratic energy function derived from the optimal damping algorithm rather than the error vector norm [46].
- Augmented Roothaan-Hall DIIS (ADIIS): Uses the quadratic augmented Roothaan-Hall energy function to obtain linear coefficients, demonstrating improved robustness compared to EDIIS for Kohn-Sham DFT [46].
- ADIIS+DIIS: Combines ADIIS and traditional DIIS, often providing highly reliable and efficient convergence [46].
The following diagram illustrates the core DIIS workflow and its integration within the SCF procedure:
Second-Order SCF (SOSCF) Methods
Theoretical Basis
Second-order SCF methods leverage Newton-Raphson techniques to achieve quadratic convergence near the solution. These approaches utilize both the orbital gradient and the orbital Hessian (second derivative) to update the orbitals [1].
The orbital rotation gradient at iteration k is given by:
gₖ = FᵏPᵏS - SPᵏFᵏ
The orbital Hessian Hₖ contains the second derivatives of the energy with respect to orbital rotations. The Newton-Raphson update step for the orbitals is obtained by solving the linear system:
HₖΔxₖ = -gₖ
where Δxₖ represents the orbital rotation step.
Practical Implementation
Exact computation and diagonalization of the orbital Hessian is computationally expensive for large systems. In practice, SOSCF implementations employ iterative subspace methods such as the co-iterative augmented Hessian (CIAH) method [1] or preconditioned conjugate gradient approaches to solve the Newton-Raphson equations approximately without explicitly constructing the full Hessian.
In PySCF, SOSCF can be invoked by decorating standard SCF objects with the newton() method [1]:
SOSCF methods typically converge in fewer iterations than DIIS, particularly near the solution where they exhibit quadratic convergence. However, each iteration is more computationally expensive due to the need to solve the Newton-Raphson equations.
Direct Minimization Methods
Fundamental Principles
Direct minimization methods approach the SCF problem as an optimization task, directly minimizing the total energy with respect to the orbital coefficients without solving the eigenvalue problem at each iteration [6]. This approach is particularly valuable for systems where DIIS struggles, such as those with small HOMO-LUMO gaps or challenging convergence characteristics.
The general direct minimization algorithm can be summarized as:
- Choose an initial set of orbitals
- Compute the energy and gradient with respect to orbital rotations
- Determine a search direction using gradient information
- Perform a line search to find the step size that minimizes the energy along this direction
- Update the orbitals and repeat until convergence
Geometric Direct Minimization
Geometric Direct Minimization (GDM) is an advanced direct minimization approach that properly accounts for the curved, hyperspherical geometry of the orbital rotation space [6]. Unlike simple steepest descent algorithms, GDM takes steps along "great circles" in this curved space, leading to significantly improved convergence properties.
In Q-Chem, GDM is the default algorithm for restricted open-shell SCF calculations and is available for all orbital types [6]. The algorithm has been extended to handle restricted open-shell cases efficiently, demonstrating superior performance compared to older direct minimization approaches [6].
Implementation and Usage
In practical implementations, direct minimization methods are often combined with DIIS in a hybrid approach. For instance, the DIIS_GDM algorithm in Q-Chem uses DIIS in the initial iterations and switches to geometric direct minimization in later stages [6]. This combination leverages the rapid early convergence of DIIS with the robustness of direct minimization near the solution.
Comparative Analysis and Performance
Algorithm Selection Guidelines
The choice of SCF algorithm depends on the specific system characteristics and computational requirements:
- DIIS is generally the preferred starting point for most molecular systems with reasonable HOMO-LUMO gaps [6].
- SOSCF methods are advantageous when high-precision convergence is required and for systems where DIIS exhibits oscillations [1].
- Direct minimization approaches are most valuable for difficult cases with small gaps, near-degeneracies, or when DIIS fails to converge [6].
Quantitative Convergence Criteria
Different quantum chemistry packages implement varying convergence thresholds. The table below summarizes standard convergence criteria in ORCA for different precision levels:
Table 1: SCF Convergence Criteria in ORCA for Different Precision Levels
| Criterion | Loose | Medium | Strong | Tight | VeryTight |
|---|---|---|---|---|---|
| TolE (Energy Change) | 1e-5 | 1e-6 | 3e-7 | 1e-8 | 1e-9 |
| TolMaxP (Max Density Change) | 1e-3 | 1e-5 | 3e-6 | 1e-7 | 1e-8 |
| TolRMSP (RMS Density Change) | 1e-4 | 1e-6 | 1e-7 | 5e-9 | 1e-9 |
| TolErr (DIIS Error) | 5e-4 | 1e-5 | 3e-6 | 5e-7 | 1e-8 |
| TolG (Orbital Gradient) | 1e-4 | 5e-5 | 2e-5 | 1e-5 | 2e-6 |
For single-point energy calculations in Q-Chem, the default convergence criterion is 10⁻⁵ atomic units for the maximum DIIS error, tightened to 10⁻⁸ for geometry optimizations and frequency calculations [45] [6].
Performance Characteristics
Table 2: Comparative Performance of SCF Convergence Algorithms
| Algorithm | Convergence Rate | Memory Requirements | Computational Cost per Iteration | Robustness for Difficult Cases |
|---|---|---|---|---|
| DIIS | Fast (near convergence) | Moderate (stores 10-20 Fock matrices) | Low | Moderate |
| EDIIS/ADIIS | Moderate to fast | Moderate (stores 10-20 Fock matrices) | Low to moderate | Good |
| SOSCF | Quadratic (near convergence) | Moderate to high | High (requires orbital Hessian) | Good |
| GDM | Linear to superlinear | Low | Moderate | Excellent |
Advanced Techniques and Troubleshooting
Initial Guess Strategies
The initial guess for the density matrix significantly impacts SCF convergence behavior. Common initialization strategies include:
- Superposition of Atomic Densities (SAD): Uses spherically averaged atomic densities or calculations to construct the initial guess [47] [1].
- Extended Hückel Theory: Provides a quantum-mechanically informed starting point that accounts for molecular structure [1].
- Core Hamiltonian: Uses eigenvectors of the one-electron core Hamiltonian, ignoring electron-electron interactions [1].
Machine learning approaches have recently emerged for generating high-quality initial guesses. Predicting electron density in a compact auxiliary basis representation using E(3)-equivariant neural networks has demonstrated significant SCF acceleration, achieving 33.3% reduction in SCF steps on systems up to 60 atoms [47].
Convergence Challenges and Solutions
Certain system classes present particular challenges for SCF convergence:
- Metallic systems with vanishing HOMO-LUMO gaps [22]
- Open-shell transition metal complexes with near-degenerate states [9]
- Antiferromagnetic systems with competing spin configurations [48]
- Systems with elongated cell dimensions causing ill-conditioning [48]
Effective strategies for addressing convergence difficulties include:
- Damping: Mixing a fraction of the previous density or Fock matrix with the new one [1].
- Level shifting: Increasing the energy of virtual orbitals to stabilize the SCF procedure [1].
- Smearing and fractional occupations: Applying finite-temperature occupation schemes or allowing fractional occupations particularly beneficial for metallic systems [1] [48].
- Mixing optimization: Adjusting mixing parameters for charge and spin densities, potentially using different mixing factors for different spin channels [48].
The following workflow provides a systematic approach for addressing SCF convergence difficulties:
Research Reagent Solutions
Table 3: Essential Computational Tools for SCF Convergence Research
| Tool Category | Specific Implementation | Function | Availability |
|---|---|---|---|
| SCF Algorithms | DIIS, ADIIS, EDIIS | Fock/density matrix extrapolation | Q-Chem, PySCF, ORCA |
| Second-Order Methods | SOSCF, CIAH Newton | Quadratic convergence acceleration | PySCF, ORCA |
| Direct Minimization | GDM, DM | Robust convergence for difficult cases | Q-Chem, ORCA |
| Initial Guess Methods | SAD, Hückel, VSAP | Starting point generation | PySCF, ORCA |
| Stability Analysis | Wavefunction stability check | Saddle point detection | PySCF, ORCA |
| Mixing Schemes | Kerker, Thomas-Fermi, Linear | Charge density mixing (periodic) | VASP, DFTK |
SCF convergence remains an active research area in electronic structure theory, with DIIS, SOSCF, and direct minimization methods representing complementary approaches with distinct strengths and limitations. DIIS excels for standard molecular systems with well-behaved convergence characteristics, while SOSCF provides high-precision convergence for challenging cases where computational resources permit. Direct minimization methods, particularly geometric direct minimization, offer robust convergence for the most difficult systems including those with small gaps and near-degeneracies.
The continuing development of hybrid approaches that intelligently switch between algorithms throughout the SCF process, combined with machine learning-enhanced initial guesses, represents the cutting edge of SCF convergence research. For computational researchers in drug development and materials science, understanding these algorithmic options and their practical implementation is crucial for efficiently tackling electronically complex systems.
The Self-Consistent Field (SCF) method represents a cornerstone in computational quantum chemistry, providing the fundamental framework for both Hartree-Fock (HF) theory and Kohn-Sham Density Functional Theory (KS-DFT). In these methodologies, the ground-state wavefunction is expressed as a single Slater determinant of molecular orbitals (MOs), and the total electronic energy is minimized subject to orbital orthogonality constraints. This approach effectively describes electrons as independent particles interacting through a mean field [1]. The SCF procedure manifests mathematically as a pseudo-eigenvalue problem known as the Roothaan-Hall equation, F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital eigenenergies [1]. The Fock matrix itself depends on the density matrix, making the equation non-linear and necessitating an iterative solution process. This article provides a comprehensive, practical guide to implementing this iterative SCF workflow within modern quantum chemistry codes, framed within broader research on optimizing this critical computational procedure.
Theoretical Foundation of the SCF Equation
At its core, the SCF method aims to solve the electronic Schrödinger equation for a many-body system by approximating the electron-electron interactions with a mean-field potential. The Fock matrix is defined as F = T + V + J + K, where T represents the kinetic energy matrix, V is the external potential (typically from nuclei), J is the Coulomb matrix, and K is the exchange matrix [1]. In KS-DFT, the exchange term is replaced by an exchange-correlation potential, but the fundamental self-consistent structure remains identical. The SCF procedure begins with an initial guess for the density or orbitals and iteratively refines this guess until the output density or Fock matrix agrees with the input to within a specified tolerance, indicating that the solution has become "self-consistent." The challenge lies in the fact that the Coulomb and exchange matrices depend on the occupied orbitals, creating a circular dependency that must be resolved through iteration [1]. Successful implementation requires careful attention to three primary phases: initial guess generation, the iterative convergence cycle, and final solution validation.
Initial Guess Generation
The starting point of an SCF calculation critically influences its convergence behavior and overall computational efficiency. A poor initial guess can lead to slow convergence, convergence to an excited state, or complete failure to converge. Fortunately, several robust initial guess strategies have been developed and implemented across quantum chemistry packages.
Table 1: Common Initial Guess Methods in SCF Calculations
| Method | Description | Typical Use Case | Implementation |
|---|---|---|---|
| Superposition of Atomic Densities (SAD) | Constructs initial density by summing densities from spin-restricted atomic calculations [1]. | Default method in many codes; good general-purpose guess. | PySCF: init_guess='atom' or 'minao' [1] |
| Core Hamiltonian ('1e') | Diagonalizes the core Hamiltonian (ignoring electron-electron interactions) [1]. | Simple fallback option; often performs poorly for molecular systems. | PySCF: init_guess='1e' [1] |
| Hückel Guess | Uses parameter-free Hückel theory based on atomic orbital energies [1]. | Improved guess over core Hamiltonian. | PySCF: init_guess='huckel' [1] |
| Chkpoint File | Reads orbitals from a previous calculation's checkpoint file [1]. | Restarting calculations or transferring between similar systems/basis sets. | PySCF: init_guess='chk' or scf.hf.from_chk() [1] |
| Atomic Potential (vSAP) | Superposition of atomic potentials evaluated on a DFT grid [1]. | DFT-specific calculations. | PySCF: init_guess='vsap' (DFT only) [1] |
For challenging systems, more advanced strategies can be employed. One powerful approach involves using the density matrix from a previous calculation on the same molecular system in a different charge or spin state, or even from a calculation with a smaller basis set, and projecting it to the target basis [1]. This is particularly useful for transition metal complexes and open-shell systems where standard guesses may fail. The initial guess can be fed directly to the SCF solver via a dm0 argument in codes like PySCF, providing maximum flexibility [1].
The SCF Iteration Cycle
Once an initial guess is established, the calculation enters the iterative cycle where the primary objective is to generate a new Fock matrix from the current density, solve for new orbitals, and construct an updated density matrix until self-consistency is achieved. The efficiency and robustness of this cycle hinge on the convergence algorithm employed.
Diagram 1: SCF Iteration Workflow. The core self-consistent field procedure involves building the Fock matrix, solving for new orbitals, and checking for convergence, with acceleration techniques applied when needed.
Convergence Algorithms
Several algorithms are available for converging the SCF procedure, each with distinct advantages and computational characteristics.
- DIIS (Direct Inversion in the Iterative Subspace): The default method in many codes, DIIS accelerates convergence by extrapolating the Fock matrix using information from previous iterations, minimizing the norm of the commutator
[F, PS]wherePis the density matrix [1]. Variants like EDIIS and ADIIS offer improved performance for certain systems [1]. - Second-Order SCF (SOSCF): Methods like the co-iterative augmented hessian (CIAH) achieve quadratic convergence by using both gradient and Hessian information [1]. These are more computationally expensive per iteration but can converge in fewer cycles, which is invoked in PySCF via
.newton()[1]. - Geometric Direct Minimization (GDM): This class of algorithms directly minimizes the total energy with respect to the orbital coefficients, often proving more robust for difficult cases, particularly for restricted open-shell calculations [49].
- Hybrid Approaches: Modern implementations often combine algorithms. For instance, Q-Chem offers
DIIS_GDM(switches from DIIS to GDM) andLS_DIIS(uses level-shifting initially then DIIS) [49]. TheROBUSTalgorithm represents a black-box workflow that automatically combines DIIS, ADIIS, and GDM with tighter thresholds [49].
Convergence Criteria and Thresholds
Determining when an SCF calculation has converged requires checking multiple criteria to ensure the solution is physically meaningful and numerically stable. Different programs offer preset levels of convergence tolerance, typically balancing computational cost against required accuracy.
Table 2: SCF Convergence Criteria in ORCA (TightSCF Example) [9]
| Criterion | Description | TightSCF Value |
|---|---|---|
| TolE | Energy change between cycles | 1e-8 Eh |
| TolMaxP | Maximum density matrix change | 1e-7 |
| TolRMSP | RMS density matrix change | 5e-9 |
| TolErr | DIIS error convergence | 5e-7 |
| TolG | Orbital gradient convergence | 1e-5 |
| TolX | Orbital rotation angle convergence | 1e-5 |
These criteria must be set compatibly with the integral evaluation threshold. In Q-Chem, the SCF_CONVERGENCE keyword sets the target for the wavefunction error (<10^-n), and the integral threshold THRESH should be set at least 3 orders of magnitude tighter [49]. For example, with SCF_CONVERGENCE = 8, THRESH should be 11 or higher [49]. The default SCF_CONVERGENCE in Q-Chem is 5 for single-point energies but 8 for geometry optimizations and frequency calculations, where higher precision is required [49].
Handling Convergence Difficulties
Despite sophisticated algorithms and careful initial guesses, SCF convergence can remain challenging for systems with small HOMO-LUMO gaps, open-shell configurations, or transition metal complexes. Several techniques can improve convergence in these problematic cases.
Technical Convergence Accelerators
- Damping: Applying damping by mixing a fraction of the previous Fock matrix with the new one (
F_new = α*F_old + (1-α)*F_new) can stabilize oscillations in the early iterations [1]. This is controlled by parameters likedampanddiis_start_cyclein PySCF, where DIIS is disabled for the first few cycles [1]. - Level Shifting: Increasing the energy of virtual orbitals artificially widens the HOMO-LUMO gap, reducing mixing between occupied and virtual spaces and stabilizing the optimization [1]. This is particularly effective for systems with near-degenerate frontier orbitals.
- Fractional Occupations and Smearing: Applying fractional occupancies according to a temperature function (smearing) helps systems with near-degenerate orbitals around the Fermi level converge by preventing occupation oscillations [1] [50]. The
Degeneratekeyword in BAND automatically smooths occupations for nearly degenerate states [50]. - Spin Breaking: For open-shell systems, initially breaking spin symmetry (
StartWithMaxSpinorVSplitin BAND) can help converge to a broken-symmetry solution [50]. TheSpinFlipoption allows targeting specific magnetic orderings [50].
Algorithmic Strategies for Problematic Cases
When standard DIIS fails, the recommended approach depends on the failure mode. If DIIS fails to find a reasonable approximate solution initially, RCA_DIIS or ADIIS_DIIS are recommended fallback options [49]. If DIIS approaches the solution but fails to converge fully, DIIS_GDM (switching to geometric direct minimization) often succeeds [49]. For systems with small HOMO-LUMO gaps where DIIS oscillates, LS_DIIS (level-shifting followed by DIIS) can be effective [49]. When these strategies fail, black-box algorithms like Q-Chem's ROBUST or ROBUST_STABLE should be tried, as they automatically combine multiple methods with appropriate thresholds [49].
Post-Convergence Analysis and Validation
Simply achieving convergence does not guarantee that the solution represents the true electronic ground state. Post-convergence analysis is essential for validating the result.
Stability Analysis
SCF solutions can sometimes converge to saddle points rather than true minima on the electronic energy landscape [1]. Stability analysis checks whether the wavefunction is stable against small perturbations. Two classes of instabilities exist [1]:
- Internal Instabilities: The solution is an excited state rather than the ground state within the same symmetry and orbital constraints.
- External Instabilities: The energy can be lowered by relaxing constraints, such as allowing restricted orbitals to become unrestricted (symmetry breaking).
Performing stability analysis after convergence is particularly important for open-shell systems, transition metal complexes, and cases where symmetry breaking is suspected. PySCF provides functionality for detecting both internal and external instabilities [1].
Property Calculation
Once a converged, stable wavefunction is obtained, various electronic properties can be computed, including:
- Dipole moments [1]
- Electronic populations (Mulliken, Lowdin, etc.)
- Electrostatic potentials
- Spectral properties (requires response theory or specialized methods like ΔSCF)
The ΔSCF method deserves special mention as it involves converging the SCF to a saddle point to model excited states, which is particularly useful for charge-transfer excitations and core-hole states where time-dependent DFT (TDDFT) may perform poorly [51]. This requires specialized convergence techniques like the Maximum Overlap Method (MOM) to maintain the desired excited state configuration throughout the iterations [51].
The Scientist's Toolkit: Essential SCF Research Reagents
Table 3: Key Computational Tools for SCF Research and Development
| Tool/Component | Function/Purpose | Implementation Examples |
|---|---|---|
| Initial Guess Generators | Provide starting density/orbitals for SCF iterations | SAD, Hückel, core Hamiltonian, checkpoint file readers [1] |
| Integral Evaluation Engines | Compute one- and two-electron integrals over basis functions | libint (ERI), libecpint (ECPs), custom GPU implementations [51] |
| DIIS Solvers | Accelerate SCF convergence via subspace extrapolation | EDIIS, ADIIS, CDIIS variants; adaptable mixing factors [1] [50] |
| Direct Minimizers | Robust convergence via energy minimization algorithms | GDM, L-BFGS; crucial for restricted open-shell and difficult cases [49] |
| Stability Analyzers | Verify solution is a true minimum, not a saddle point | Internal/external stability checks; follow-up wavefunction optimization [1] |
| Implicit Solvent Models | Incorporate solvent effects into the SCF procedure | PCM, CPCM, SMD, SMx models; requires additional potential terms [51] |
| Dispersion Corrections | Add non-local correlation effects to DFT | D3, D4 corrections; available as external potentials [51] |
| Wavefunction I/O Handlers | Store/retrieve orbitals and densities for restarting | Checkpoint files; basis set projection for transfer between calculations [1] |
The Self-Consistent Field method remains the computational backbone of modern quantum chemistry, forming the foundation for both sophisticated wavefunction methods and practical density functional calculations. A robust SCF implementation requires careful integration of multiple components: intelligent initial guesses, efficient Fock matrix construction, sophisticated convergence accelerators, and thorough post-convergence validation. The ongoing development of improved initial guesses (potentially leveraging machine learning), more robust convergence algorithms, and better implicit solvent models represents active research frontiers that will further enhance the reliability and applicability of SCF methods [51]. For researchers implementing these methods, understanding the interplay between different algorithmic choices and system characteristics is paramount for success across diverse chemical spaces, from drug-like organic molecules to challenging transition metal catalysts and excited state systems.
Basis Set Selection and Impact on SCF Convergence and Accuracy
The self-consistent field (SCF) method is the foundational algorithm for solving electronic structure problems in both Hartree-Fock theory and Kohn-Sham density functional theory [1]. In this iterative procedure, the selection of an appropriate basis set—the set of mathematical functions used to represent molecular orbitals—is a critical decision that directly governs the accuracy of computed properties and the feasibility of the calculation itself [52]. The basis set determines how the electronic wavefunction is represented, influencing everything from total energies and molecular geometries to reaction barriers and spectroscopic properties [52] [53].
This technical guide examines the intricate relationship between basis set selection, SCF convergence behavior, and ultimate calculation accuracy within the context of SCF iterative process research. For computational chemists and drug development professionals, understanding these relationships is essential for designing efficient computational protocols that balance numerical precision with practical computational constraints. We present systematic methodologies for basis set selection, quantitative performance comparisons, and practical protocols for overcoming SCF convergence challenges, particularly for complex systems such as transition metal complexes and large organic molecules relevant to pharmaceutical research.
Basis Set Fundamentals and Hierarchy
Basis Set Composition and Types
In electronic structure calculations, basis sets consist of atom-centered functions that approximate molecular orbitals. The most common types are:
- Numerical Atomic Orbitals (NAOs): Obtained by numerically solving Kohn-Sham equations for isolated spherical atoms [52]
- Slater-Type Orbitals (STOs): Exponential functions that accurately represent atomic orbitals but require more computational resources [52]
- Gaussian-Type Orbitals (GTOs): The most widely used functions in quantum chemistry software due to computational efficiency, with modern variants including polarization, diffuse, and correlation-consistent functions [52] [54]
Most general-purpose quantum chemistry packages utilize GTOs, while specialized software like ADF employs STOs. The choice between these types involves trade-offs between accuracy, computational efficiency, and numerical stability [54].
Basis Set Hierarchy and Nomenclature
Basis sets follow a well-defined hierarchy based on their completeness and accuracy. The standard classification includes:
- Single Zeta (SZ): Minimal basis sets with one basis function per atomic orbital; computationally efficient but often inaccurate for quantitative predictions [52]
- Double Zeta (DZ): Two basis functions per atomic orbital; offer improved accuracy with moderate computational cost [52]
- Triple Zeta (TZ): Three basis functions per atomic orbital; provide good balance between accuracy and computational cost [52]
- Quadruple Zeta (QZ): Four basis functions per atomic orbital; deliver high accuracy for benchmarking [52]
Polarization functions (denoted by 'P') account for orbital deformation in molecular environments, while diffuse functions (often denoted by 'aug-' prefix) improve the description of electron-dense regions such as anions and excited states [54]. The systematic improvement from SZ to QZ basis sets represents increasing computational cost and accuracy, with each step providing better approximation to the complete basis set (CBS) limit [52].
Table 1: Standard Basis Set Hierarchy and Characteristics
| Basis Set | Zeta Quality | Polarization Functions | Typical Use Case |
|---|---|---|---|
| SZ | Single | None | Quick test calculations |
| DZ | Double | None | Pre-optimization |
| DZP | Double | Single | Geometry optimizations of organic systems |
| TZP | Triple | Single | Recommended balanced choice |
| TZ2P | Triple | Double | Accurate description of virtual orbitals |
| QZ4P | Quadruple | Quadruple | Benchmarking |
Quantitative Impact of Basis Set Selection
Accuracy Versus Computational Cost
The choice of basis set represents a fundamental trade-off between computational cost and accuracy. Larger basis sets provide better approximation to the complete basis set limit but demand significantly more computational resources [52].
Table 2: Basis Set Performance Comparison for a (24,24) Carbon Nanotube [52]
| Basis Set | Energy Error (eV/atom) | CPU Time Ratio (Relative to SZ) |
|---|---|---|
| SZ | 1.8 | 1.0 |
| DZ | 0.46 | 1.5 |
| DZP | 0.16 | 2.5 |
| TZP | 0.048 | 3.8 |
| TZ2P | 0.016 | 6.1 |
| QZ4P | Reference | 14.3 |
As demonstrated in Table 2, moving from SZ to DZP reduces the energy error by nearly 92% while increasing computational time by only 2.5 times. However, further improvements to QZ4P come at substantially higher computational cost (14.3×) for diminishing returns in accuracy [52]. This nonlinear relationship underscores the importance of selecting basis sets appropriate for the target property.
Property-Specific Basis Set Dependencies
Different molecular properties converge at varying rates with respect to basis set size:
- Total Energies: Show systematic improvement with larger basis sets but require high zeta-levels for chemical accuracy [52]
- Energy Differences: Reaction energies and barrier heights benefit from error cancellation, often converging with smaller basis sets [52]
- Molecular Geometries: Generally converge at double-zeta or triple-zeta levels with polarization functions [54]
- Band Gaps: Require polarization functions for adequate description of virtual orbitals; DZ often proves inadequate while TZP captures trends effectively [52]
For properties dependent on electron density tail regions, such as weak intermolecular interactions or anion properties, diffuse functions become essential [54]. However, these same functions can introduce numerical instability and SCF convergence difficulties [55].
Basis Set Selection Methodology
Systematic Selection Framework
The following diagram illustrates a logical decision workflow for basis set selection, balancing accuracy requirements, system characteristics, and computational constraints:
Recommended Basis Sets by Application
For different computational scenarios, the following basis sets represent current best practices:
- General Organic Molecules: def2-TZVP or TZP offers the optimal balance between accuracy and computational cost [52] [56]
- Transition Metal Complexes: def2-TZVP with appropriate effective core potentials [56] [57]
- Weak Interactions and Anions: aug-cc-pVTZ or other diffuse-augmented basis sets [54]
- Benchmark Calculations: QZ4P or aug-cc-pVQZ for approaching the complete basis set limit [52]
- Geometry Optimizations: DZP or def2-SVP provides reasonable results with efficiency [52]
The def2 basis sets from Karlsruhe are particularly recommended for DFT calculations as they are available for the entire periodic table and include effective core potentials for heavy elements [56]. For correlated methods beyond DFT, such as coupled-cluster theory, the Dunning cc-pVXZ series is preferred due to their systematic convergence to the complete basis set limit [56] [54].
SCF Convergence Challenges and Solutions
Basis Set Specific Convergence Issues
The SCF iterative process can encounter significant convergence challenges with certain basis set types:
- Large Basis Sets: QZ4P and larger basis sets often exhibit convergence difficulties due to increased linear dependencies and poorer condition numbers [58]
- Diffuse Functions: Augmented basis sets (e.g., aug-cc-pVXZ) frequently cause convergence problems, particularly for systems with small HOMO-LUMO gaps [55]
- Basis Set Superposition Error (BSSE): Inadequate basis set size can lead to BSSE, where subsystems artificially borrow basis functions from neighboring atoms, affecting both intermolecular and intramolecular interactions [53]
The intramolecular BSSE is particularly problematic as it affects all calculations with limited basis sets, not just weakly interacting complexes, and can lead to unphysical molecular geometries and reaction energies [53].
Comprehensive SCF Convergence Protocol
The following workflow provides a systematic approach for addressing SCF convergence problems:
Specialized Techniques for Difficult Systems
For particularly challenging cases, specialized approaches are necessary:
Transition Metal Complexes and Open-Shell Systems:
- Use
SlowConvorVerySlowConvkeywords to increase damping [57] - Employ KDIIS algorithm with SOSCF for faster convergence [57]
- Delay SOSCF start to smaller gradient thresholds (e.g., 0.00033 instead of default 0.0033) [57]
- For pathological cases, increase DIIS expansion vectors to 15-40 and reduce direct reset frequency [57]
Systems with Diffuse Functions:
- Enable full Fock matrix rebuilds (
directresetfreq 1) to reduce numerical noise [57] - Start SOSCF earlier in the convergence process [57]
- Consider removing diffuse functions for initial convergence, then re-adding for final energy evaluation
Linear Dependency Issues:
- For large or diffuse basis sets, remove linearly dependent basis functions [57]
- Increase integration grid accuracy to reduce numerical noise [58] [57]
- Adjust cutoff values to accommodate large basis set exponents [58]
Research Reagent Solutions
Table 3: Essential Computational Tools for Basis Set and SCF Research
| Research Reagent | Function | Application Context |
|---|---|---|
| def2 Basis Set Series | Balanced GTO basis sets for entire periodic table | General-purpose DFT calculations for organic and inorganic systems |
| cc-pVXZ Series | Correlation-consistent basis sets for systematic CBS extrapolation | High-accuracy wavefunction theory calculations |
| MOLOPT Basis Sets | Optimized for condensed phase with numerical stability constraints | Molecular dynamics and periodic systems |
| DIIS Algorithm | Extrapolates Fock matrix from previous iterations | Standard SCF acceleration |
| SOSCF | Second-order convergence with quadratic convergence near solution | Difficult cases where DIIS fails |
| Level Shifting | Artificially increases HOMO-LUMO gap | Systems with small gaps or near-degeneracies |
| Electron Smearing | Fractional occupancies for near-degenerate levels | Metallic systems and small-gap semiconductors |
| Counterpoise Correction | Corrects for Basis Set Superposition Error | Non-covalent interaction energies |
Experimental Protocols for Basis Set Assessment
Protocol 1: Basis Set Convergence Study
Objective: Determine the optimal basis set for a specific molecular property while balancing computational cost and accuracy.
Methodology:
- Select a series of basis sets along the zeta hierarchy (e.g., DZP → TZP → TZ2P → QZ4P)
- Perform single-point energy calculations on a representative molecular system
- Calculate the target property (formation energy, reaction energy, band gap) with each basis set
- Compute relative errors using the largest basis set as reference
- Compare computational costs (CPU time, memory requirements)
Interpretation: Identify the point where property changes become smaller than desired accuracy thresholds, indicating the optimal basis set for future calculations [52].
Protocol 2: SCF Stability Analysis
Objective: Ensure the converged SCF solution represents a true minimum rather than a saddle point.
Methodology:
- After SCF convergence, perform stability analysis following established procedures [1]
- Check for internal instabilities (convergence to excited states)
- Check for external instabilities (lower energy solutions with different symmetry constraints)
- If unstable states are detected, employ more robust convergence methods or modify initial guess
Interpretation: Stable solutions maintain the same electronic state under small perturbations, while unstable solutions may collapse to lower-energy configurations with different symmetry [1].
Protocol 3: Intramolecular BSSE Assessment
Objective: Quantify the effect of intramolecular basis set superposition error on molecular properties.
Methodology:
- Select a series of chemically similar molecules of increasing size (e.g., hydrocarbons)
- Compute target properties (e.g., proton affinities) with multiple basis sets
- Analyze the deviation from experimental values as a function of basis set size and molecular size
- Compare with reference calculations at the complete basis set limit where available
Interpretation: Significant variations with basis set size that correlate with molecular size indicate substantial intramolecular BSSE, necessitating larger basis sets or correction schemes [53].
Basis set selection remains a critical consideration in SCF-based electronic structure calculations, directly influencing both the accuracy of computed properties and the robustness of the SCF iterative process. The hierarchical nature of basis sets provides a systematic pathway for approaching the complete basis set limit, but practical calculations require careful balance between numerical accuracy and computational feasibility.
For most applications involving drug development and molecular design, triple-zeta basis sets with polarization functions (TZP, def2-TZVP) offer the optimal balance, providing chemical accuracy with reasonable computational cost. For specialized properties involving weak interactions, excited states, or transition metal complexes, targeted basis set selection with appropriate convergence protocols becomes essential.
The interplay between basis set selection and SCF convergence behavior necessitates an integrated approach to computational protocol development, where basis set choice is considered alongside convergence acceleration techniques and initial guess strategies. By adopting the systematic methodologies outlined in this guide, researchers can develop more robust and reliable computational workflows for pharmaceutical applications and molecular design.
The Self-Consistent Field (SCF) iterative procedure is a cornerstone of computational quantum chemistry, enabling the determination of electronic wavefunctions and energies for molecular systems. The reliability and efficiency of this process are highly dependent on the electronic structure of the system under investigation. Systems are broadly categorized into three classes based on their electronic configurations: closed-shell, open-shell, and multiconfigurational. Closed-shell systems, characterized by all electrons being paired in doubly occupied orbitals, represent the simplest case and are typically well-described by a single Slater determinant using the Restricted Hartree-Fock (RHF) method. In contrast, open-shell systems contain unpaired electrons and require more sophisticated treatments such as Unrestricted Hartree-Fock (UHF) or Restricted Open-Shell Hartree-Fock (ROHF) to properly account for spin polarization. The most challenging cases are multiconfigurational systems, where a single determinant is insufficient to describe the electronic structure due to near-degeneracy effects or bond-breaking situations. These systems necessitate advanced methods like Multi-Configurational SCF (MCSCF), particularly the Complete Active Space SCF (CASSCF) approach, which provides a conceptually sound framework for capturing static correlation by allowing multiple electronic configurations within an active orbital space [59] [60].
Understanding the distinctions between these system types is crucial for researchers and drug development professionals who utilize quantum chemical calculations. The choice of method directly impacts the accuracy of predicting molecular properties, reaction pathways, and intermolecular interactions relevant to pharmaceutical development. As quantum techniques continue to offer glimpses of next-generation medicine through more accurate molecular simulations [61], properly handling these different system types becomes increasingly important for accelerating drug discovery and materials design.
Theoretical Foundations of System Classifications
Electronic Structure Characteristics
The fundamental differences between closed-shell, open-shell, and multiconfigurational systems originate from their distinct electron occupancy patterns and spin characteristics:
Closed-Shell Systems: These systems exhibit a singlet ground state with total spin quantum number S=0. All molecular orbitals are either doubly occupied or empty, resulting in a wavefunction that can be adequately described by a single determinant. The spatial part of the α- and β-spin orbitals are identical in the RHF formalism, which reduces computational complexity. Such systems are typically encountered in organic molecules where all electrons are paired, such as methane, ethane, and most stable organic compounds in their ground states.
Open-Shell Systems: Characterized by the presence of unpaired electrons, these systems have non-zero spin quantum numbers (S>0). They can be further classified into:
- High-spin systems: Where unpaired electrons maximize total spin (e.g., triplet oxygen, transition metal complexes)
- Radical species: With one or more unpaired electrons (e.g., methyl radical, nitric oxide)
- Spin-polarized systems: Where different spin populations exist in different molecular regions
The UHF approach allows α- and β-spin orbitals to have different spatial distributions, providing flexibility but potentially introducing spin contamination [6].
Multiconfigurational Systems: These challenging cases arise when multiple electronic configurations contribute significantly to the wavefunction, making a single-determinant description qualitatively incorrect. This occurs in:
- Bond-breaking processes: Where the Hartree-Fock method fails dissociating bonds correctly [59]
- Near-degenerate systems: Where molecular orbitals are close in energy
- Transition metal complexes: With multiple nearly-degenerate d-orbitals
- Diradicals and polyradicals: With significant non-dynamic electron correlation
The MCSCF method addresses these challenges by employing a linear combination of configuration state functions to approximate the exact electronic wavefunction [59].
Mathematical Formulations
The mathematical description of these systems varies significantly in complexity:
For closed-shell systems, the RHF method minimizes the energy functional: [ E{\text{RHF}} = 2\sumi^{N/2} H{ii} + \sum{ij}^{N/2} [2J{ij} - K{ij}] ] where (H{ii}) are core Hamiltonian elements, and (J{ij}) and (K_{ij}) are Coulomb and exchange integrals, respectively.
Open-shell systems require more complex formulations. The UHF method employs different spatial orbitals for α and β spins: [ E{\text{UHF}} = \sumi^\alpha H{ii} + \sumi^\beta H{ii} + \frac{1}{2}\sum{ij}^\alpha [J{ij}^\alpha - K{ij}^\alpha] + \frac{1}{2}\sum{ij}^\beta [J{ij}^\beta - K{ij}^\beta] + \sum{ij}^{\alpha\beta} J_{ij}^{\alpha\beta} ]
Multiconfigurational systems utilize the MCSCF wavefunction ansatz: [ \Psi{\text{MCSCF}} = \sumI CI \PhiI ] where (\PhiI) are configuration state functions (CSFs) or Slater determinants, and both the CI coefficients (CI) and molecular orbital coefficients are optimized self-consistently [59]. In the CASSCF variant, a full configuration interaction (FCI) expansion within a selected active space ensures a balanced treatment of static correlation.
Table 1: Key Characteristics of Different Electronic System Types
| System Type | Spin Character | Determinant Requirement | Key Methods | Typical Applications |
|---|---|---|---|---|
| Closed-Shell | S=0, all electrons paired | Single determinant | RHF, RKS-DFT | Stable organic molecules, saturated compounds |
| Open-Shell | S>0, unpaired electrons | Single determinant (but different for α/β spins) | UHF, ROHF, UKS-DFT | Radicals, transition metal complexes, triplet states |
| Multiconfigurational | Variable, often open-shell | Multiple determinants | MCSCF, CASSCF, RASSCF | Bond breaking, diradicals, excited states, transition states |
Computational Approaches and Protocols
Initial Setup and Wavefunction Specification
Proper initialization is crucial for successful SCF convergence across all system types:
For closed-shell systems, the initialization is typically straightforward. Most quantum chemistry packages automatically detect closed-shell systems based on electron count and spin multiplicity. The default Aufbau orbital occupation usually provides a good starting point for the SCF procedure [62].
For open-shell systems, explicit specification of the wavefunction is often necessary. In Molpro, for instance, researchers are recommended to use WF, OCC, CLOSED, or OPEN directives to uniquely define the wavefunction when the default occupation guesses are incorrect [62]. Specifying the correct spin symmetry is essential to avoid convergence to unphysical solutions.
For multiconfigurational systems, the active space selection represents the most critical step. The CASSCF method requires specification of the number of active electrons and orbitals [60]. As highlighted in the PySCF documentation, "Hartree-Fock orbitals are often poor for systems with significant static correlation. In such cases, orbitals from density functional calculations often yield better starting points for CAS calculations" [60]. The automated AVAS and DMET_CAS procedures can assist in active space selection by identifying important orbitals based on atomic orbital projections [60].
SCF Convergence Algorithms and Strategies
Different system types require specialized convergence algorithms:
Closed-shell systems typically converge efficiently with the standard Direct Inversion in the Iterative Subspace (DIIS) method [6] [9]. DIIS accelerates convergence by extrapolating Fock matrices from previous iterations using error vectors derived from the commutator of the Fock and density matrices [6].
Open-shell systems, particularly those with significant spin polarization or near-degeneracy, often present convergence challenges. For these cases, Geometric Direct Minimization (GDM) has emerged as a highly robust alternative [6]. GDM properly accounts for the curved geometry of orbital rotation space, leading to more reliable convergence. As noted in the Q-Chem manual, "GDM is the default algorithm for restricted open-shell SCF calculations and is available for all orbital types" [6].
Multiconfigurational systems require specialized MCSCF convergence protocols. The second-order convergence methods, which approximate the orbital Hessian, typically offer the most efficient approach [60]. For challenging cases, state-averaged MCSCF can improve convergence by simultaneously optimizing multiple states.
Table 2: Recommended Convergence Algorithms for Different System Types
| System Type | Primary Algorithm | Fallback Algorithm | Key Parameters |
|---|---|---|---|
| Closed-Shell | DIIS | DIISGDM, RCADIIS | DIISSUBSPACESIZE=15, SCF_CONVERGENCE=8 |
| Open-Shell | GDM | DIIS_GDM, ADIIS | SCFALGORITHM=GDM, MAXSCF_CYCLES=100 |
| Multiconfigurational | Second-order MCSCF | Single-state MCSCF | MAXCYCLEMCSCF=50, LEVEL_SHIFT=0.5 |
Practical Convergence Protocols
Implementing robust convergence protocols requires careful parameter selection:
For standard closed-shell calculations, the default DIIS algorithm with a subspace size of 15-20 and energy convergence criterion of 10(^{-8}) a.u. typically suffices [6] [9]. For geometry optimizations and frequency calculations, tighter convergence (10(^{-10}) to 10(^{-12}) a.u.) may be necessary.
Open-shell systems benefit from GDM with the following ORCA settings for challenging cases [9]:
Increasing the maximum number of SCF cycles to 100-200 is recommended for transition metal complexes [6] [9].
Multiconfigurational systems require careful active space selection and convergence monitoring. The CASSCF implementation in PySCF supports various strategies for active space selection, including [60]:
- Manual selection based on molecular orbital inspection
- Automated selection using natural orbitals from MP2 or CISD calculations
- Projection-based methods (AVAS) that select orbitals based on overlap with specific atomic orbitals
Advanced Methods for Challenging Systems
Multiconfigurational Methods: MCSCF and CASSCF
Multiconfigurational self-consistent field (MCSCF) methods provide the most general framework for treating systems where a single determinant description fails. The Complete Active Space SCF (CASSCF) method, a specific variant of MCSCF, divides the molecular orbitals into three sets: inactive (doubly occupied), active (partially occupied), and virtual (unoccupied) [59] [60]. A full configuration interaction (FCI) calculation is then performed within the active space, while the orbital coefficients for all orbitals are optimized simultaneously.
The CASSCF wavefunction is defined as: [ \Psi{\text{CASSCF}} = \sum{I} CI \PhiI^{\text{active}} \otimes \Phi^{\text{inactive}} \otimes \Phi^{\text{virtual}} ] where the sum runs over all possible distributions of active electrons among active orbitals.
For larger systems where full CASSCF becomes computationally prohibitive, the Restricted Active Space SCF (RASSCF) method offers a practical alternative by restricting the number of electrons in certain orbital subspaces [59]. RASSCF allows single and double excitations from specific subsets of orbitals while limiting the maximum number of electrons in others, significantly reducing the computational cost while maintaining accuracy for many chemical applications.
Density Matrix Renormalization Group (DMRG)
For strongly correlated systems with large active spaces, the Density Matrix Renormalization Group (DMRG) has emerged as a powerful alternative to traditional CI methods [63]. DMRG is a numerical variational technique that efficiently obtains the low-energy physics of quantum many-body systems with high accuracy. The algorithm attempts to find the lowest-energy matrix product state wavefunction of a Hamiltonian [63].
The key advantage of DMRG lies in its ability to handle active spaces far beyond the limits of conventional CASSCF, making it particularly valuable for studying complex transition metal clusters, large polycyclic aromatic hydrocarbons, and multinuclear catalytic systems. DMRG can be combined with CASSCF in the form of DMRG-CASSCF, where DMRG serves as the configuration interaction solver within the active space.
Quantum Computing Approaches
The emerging field of quantum computing offers promising long-term solutions for electronic structure problems that challenge classical computational methods. Quantum computers can naturally capture the behavior of molecules at a fundamental level, representing and manipulating many possible states of a system simultaneously [61]. This capability allows quantum computers to simulate molecular interactions with extremely high accuracy in principle.
Current research has demonstrated hybrid quantum-classical approaches for molecular problems. As highlighted in recent studies, "hybrid techniques can tackle small, well-defined molecular problems more efficiently than either approach alone" [61]. Companies like QSimulate, Qubit Pharmaceuticals, and Gero are exploring practical applications of these techniques for drug discovery, leveraging quantum-inspired algorithms even on classical hardware [61].
In the pharmaceutical industry, McKinsey estimates potential value creation of $200 billion to $500 billion by 2035 through quantum computing applications in life sciences [64]. Major companies including AstraZeneca, Boehringer Ingelheim, and Merck KGaA are actively exploring quantum computing for molecular simulations [64].
Successful computational studies of diverse molecular systems require familiarity with specialized tools and techniques:
Table 3: Research Reagent Solutions for Electronic Structure Calculations
| Tool/Resource | System Type | Function | Implementation Examples |
|---|---|---|---|
| DIIS Algorithm | Closed-Shell, Open-Shell | Convergence acceleration | Default in Q-Chem, ORCA, PySCF |
| GDM Algorithm | Open-Shell, Difficult Cases | Robust convergence | Q-Chem (default for ROHF), ORCA |
| CASSCF | Multiconfigurational | Static correlation treatment | PySCF, ORCA, Molpro, OpenMolcas |
| DMRG | Strongly Correlated Systems | Large active space calculations | PySCF (with external solvers), Block |
| AVAS Procedure | Multiconfigurational | Automated active space selection | PySCF (pyscf.mcscf.avas) |
| Natural Orbitals | Multiconfigurational | Improved orbital initialization | MP2/CISD natural orbitals in PySCF |
| Stability Analysis | All Systems | Verification of solution quality | ORCA (! STABLE keyword) |
| Density Fitting | All Systems | Computational acceleration | DF-HF in Molpro, RI-J in ORCA |
Visualization and Analysis Tools
Orbital visualization is crucial for multiconfigurational methods, as emphasized in the PySCF documentation: "We always advise users to visualize their chosen active orbitals before starting large/expensive calculations" [60]. Recommended tools include:
- JMol: User-friendly molecular viewer suitable for less experienced researchers
- Molden: Comprehensive package for displaying molecular orbitals and electron densities
- VMD: Advanced visualization with extensive scripting capabilities
These tools help researchers verify that selected active spaces correspond to chemically intuitive orbitals (e.g., d-orbitals in transition metals, π-orbitals in conjugated systems).
Specialized Software Packages
Different quantum chemistry packages offer unique strengths for specific system types:
- PySCF: Excellent for method development and multiconfigurational calculations with Python API [60]
- ORCA: Robust open-shell and transition metal calculations with user-friendly input [9]
- Q-Chem: Advanced algorithms for difficult SCF convergence [6]
- Molpro: High-accuracy multireference methods with efficient implementation [62]
- WIEN2k: Specialized for solid-state systems with advanced diagonalization methods [65]
Applications in Drug Discovery and Materials Science
The proper handling of different electronic system types has profound implications for pharmaceutical research and materials development:
Drug Discovery Applications
In drug discovery, accurate molecular simulations can significantly reduce the time and cost associated with bringing new therapies to patients [64]. Specific applications include:
Protein-Ligand Binding: Quantum mechanical calculations provide accurate binding energies, particularly for metalloenzymes where multiconfigurational character is common. Boehringer Ingelheim's collaboration with PsiQuantum focuses on calculating electronic structures of metalloenzymes critical for drug metabolism [64].
Reaction Pathway Analysis: Studying drug metabolism pathways often involves modeling bond-breaking processes that require multiconfigurational methods. Hybrid quantum-classical approaches have been successfully applied to model prodrug activation and covalent inhibitors targeting cancer-associated mutations [61].
Solvation Effects: Understanding how proteins adopt different geometries in solvent environments is vital for drug targeting. Quantum computers can accurately model these effects, especially for orphan proteins with limited experimental data [64].
Toxicity Prediction: Identifying potential off-target effects through precise reverse docking simulations helps reduce late-stage failures in drug development [64].
Emerging Methodologies
The integration of quantum techniques with AI represents a promising frontier. As noted by Toru Shiozaki of QSimulate, "As AI continues to evolve, quantum mechanics will be more important than ever in the next frontier of drug discovery where both quantum and AI technologies will act as complementary forces" [61]. Quantum-informed AI can explore chemical spaces not accessible to classical methods, generating novel molecular structures with desired properties.
Companies are already demonstrating practical applications: Gero has combined machine learning with quantum components to generate thousands of novel drug-like molecules not present in training datasets [61]. Qubit Pharmaceuticals uses quantum-accurate data to train AI models that simulate dynamic protein-drug interactions at unprecedented scales [61].
The sophisticated handling of closed-shell, open-shell, and multiconfigurational systems within the SCF framework represents an essential capability for modern computational chemistry in pharmaceutical research and materials design. While closed-shell systems typically yield to standard protocols, open-shell species require specialized algorithms like GDM for robust convergence, and multiconfigurational cases demand advanced methods such as CASSCF with careful active space selection. The emerging integration of quantum computing approaches with classical methods promises to further extend the boundaries of simulatable systems, potentially revolutionizing drug discovery processes. As computational power increases and algorithms refine, the systematic application of these specialized techniques will continue to enhance our ability to model complex molecular systems with pharmaceutical relevance, ultimately accelerating the development of novel therapeutic agents.
The Complete Active Space Self-Consistent Field (CASSCF) method represents a cornerstone of quantum chemistry for treating systems where the single-configuration picture of Hartree-Fock theory fails. As a multiconfigurational and multireference method, CASSCF provides a qualitatively correct wavefunction for studying molecules with significant static correlation effects, such as open-shell transition metal complexes, bond-breaking processes, and electronically excited states [66] [67]. Within the broader context of Self-Consistent Field (SCF) iterative processes, CASSCF extends the fundamental variational principle by optimizing both the molecular orbital coefficients and the configuration interaction (CI) coefficients simultaneously [66]. This capability makes it an indispensable tool for researchers and drug development professionals investigating catalytic mechanisms, photochemical reactions, and other complex processes involving transition metals and radical species where electron correlation plays a decisive role.
Theoretical Foundation of CASSCF
The CASSCF Wavefunction and Energy Functional
The CASSCF wavefunction, (\left| \PsiI^S \right\rangle), for a state (I) with total spin (S) is expressed as a linear combination of configuration state functions (CSFs) [66]: [ \left| \PsiI^S \right\rangle= \sum{k} { C{kI} \left| \Phik^S \right\rangle} ] These CSFs ((\left| \Phik^S \right\rangle)) are spin-adapted linear combinations of Slater determinants. The method involves two sets of variational parameters: the CI coefficients ((C{kI})) that weight each CSF, and the molecular orbital coefficients ((c{\mu i})) that expand the MOs in the atomic basis set [66]. The energy, [ E\left({ \mathrm{\mathbf{c} },\mathrm{\mathbf{C} }} \right)=\frac{\left\langle { \Psi {I}^{S} \left|{ \hat{{H} }{\text{BO} } } \right|\Psi{I}^{S} } \right\rangle}{\left\langle { \Psi{I}^{S} \left|{ \Psi_{I}^{S} } \right.} \right\rangle}, ] is variational and provides an upper bound to the true total energy. A crucial conceptual point is that CASSCF is designed not for quantitative accuracy but for qualitative correctness, providing a robust starting point for dynamic correlation treatments [66].
Orbital Spaces and the Active Electron Concept
A fundamental operational step in CASSCF is the partitioning of the molecular orbital space into three distinct subspaces [66]:
- Inactive Orbitals: Doubly occupied in all CSFs.
- Active Orbitals: Orbitals with variable occupation across the CSFs.
- External Orbitals: Unoccupied (virtual) orbitals in all CSFs.
The active space is defined by the number of active electrons ((n)) distributed among the active orbitals ((m)), denoted as CASSCF((n),(m)). Within this active space, a full configuration interaction calculation is performed, thereby capturing static correlation [66]. The wavefunction and energy remain invariant to unitary transformations within each of these three subspaces.
State-Averaging for Multiple Electronic States
For studying multiple electronic states simultaneously, CASSCF employs the state-averaging (SA-CASSCF) approach. Instead of optimizing for a single state, the orbitals are optimized for an average of several states using user-defined weights ((wI)) that sum to unity [66]. The averaged one- and two-particle density matrices are computed as: [ \Gamma{q}^{p\left({ av} \right)} =\sum\limitsI { w{I} \Gamma {q}^{p\left( I \right)} } \quad \text{and} \quad \Gamma{qs}^{pr\left({ av} \right)} =\sum\limitsI { w{I} \Gamma_{qs}^{pr\left( I \right)} } ] This yields the average energy functional. State-averaging is particularly crucial for calculating excited states and for ensuring a consistent description of potential energy surfaces across state crossings [66].
CASSCF Protocol and Workflow
The successful execution of a CASSCF calculation requires careful attention to the active space selection and the convergence of the SCF procedure. The following workflow diagram outlines the key steps and decision points.
Active Space Selection Protocol
The choice of the active space is the most critical step in a CASSCF study, directly influencing the qualitative accuracy of the wavefunction. The table below summarizes common strategies and their applications.
Table: Protocols for Active Space Selection in CASSCF Calculations
| Protocol | Description | Typical Application |
|---|---|---|
| Chemical Intuition | Selection based on known reactive orbitals, valence orbitals, and broken bonds. | Prototypical organic diradicals, first-row transition metal complexes. |
| Automatic Selection | Uses algorithms based on natural orbital occupations, orbital entanglement, or machine learning [67]. | Complex systems where manual selection is difficult; high-throughput studies. |
| Weighted Active Space (WASP) | A systematic protocol for consistent active space assignment across uncorrelated geometries [67]. | Machine-learned potential energy surfaces; reaction dynamics. |
A well-chosen active space should ideally consist of orbitals with occupation numbers between approximately 0.02 and 1.98 to ensure robust convergence [66]. Orbitals with occupations too close to 2.0 or 0.0 can lead to convergence difficulties as the energy becomes weakly dependent on rotations involving these orbitals.
Convergence and Optimization Techniques
CASSCF optimizations are more complex than single-reference SCF due to strong coupling between CI and orbital coefficients and the presence of many local minima [66]. Two primary classes of algorithms are used:
- First-Order Methods: Require only a small subset of transformed integrals, making them less computationally demanding and suitable for larger systems [66].
- Second-Order Methods: Utilize the Newton-Raphson approach, which involves solving an eigenvalue problem with the orbital Hessian [66] [68]: [ \begin{pmatrix} 0 & \mathbf{g} \ \mathbf{g} & \mathbf{H} \end{pmatrix} \begin{pmatrix} 1 \ \mathbf{t} \end{pmatrix} = \varepsilon \begin{pmatrix} 1 \ \mathbf{t} \end{pmatrix} ] Here, (\mathbf{g}) is the orbital gradient and (\mathbf{H}) is the orbital Hessian. These methods exhibit quadratic convergence but require a larger set of integrals, limiting their application to smaller molecules [66].
Practical Application and Research Toolkit
The Scientist's Toolkit: Essential Reagents and Software
Table: Key "Research Reagent Solutions" for CASSCF Studies
| Item / Resource | Function / Role in CASSCF |
|---|---|
| High-Quality Basis Sets | Provide the mathematical basis for expanding molecular orbitals (e.g., correlation-consistent cc-pVXZ, ANO-type basis sets). |
| Initial Orbital Guess | A critical starting point (e.g., from ROHF, UHF, or fragment calculations) to guide convergence to the desired state. |
| Automated Active Space Solvers | Algorithms (e.g., based on natural orbital occupations or entanglement) to assist in selecting the active orbitals [67]. |
| CI Solver | The computational engine that solves the full-CI problem within the active space (e.g., DMRG for very large active spaces) [66]. |
| Post-CASSCF Methods | Techniques like CASPT2, NEVPT2, or MC-PDFT that add dynamic correlation for quantitative energy accuracy [67]. |
Quantitative Benchmarks: The HNO Molecule Case
The application of CASSCF to the HNO molecule, as presented in the seminal 1981 paper, serves as a classic benchmark [68]. The table below compares calculated properties against experimental values, demonstrating the method's capabilities and limitations.
Table: CASSCF Performance for Ground State HNO Geometry and Excitation Energies [68]
| Property | CASSCF Result | Experimental Value |
|---|---|---|
| R(NO) (Å) | 1.215 | 1.212 |
| R(NH) (Å) | 1.079 | 1.063 |
| ∠HNO (°) | 108.8 | 108.6 |
| Excitation Energy to ³A″ (eV) | 0.67 | 0.85 |
| Excitation Energy to ¹A″ (eV) | 1.52 | 1.63 |
The results show that CASSCF provides excellent equilibrium geometries but tends to overestimate excitation energies, a shortcoming attributed to the lack of dynamic electron correlation outside the active space [68].
Advanced Protocols: Multireference Machine Learning Potentials
A cutting-edge application of CASSCF is in generating data for training machine learning potentials (MLPs). This faces a unique challenge: ensuring a consistent active space across diverse, uncorrelated molecular geometries sampled during active learning [67]. The Weighted Active Space Protocol (WASP) was developed to address this. WASP provides a systematic, geometry-independent rule for active orbital assignment, enabling the consistent labeling of multireference data needed to train MLPs without introducing spurious discontinuities from varying active spaces [67]. When integrated with methods like multiconfiguration pair-density functional theory (MC-PDFT), this protocol allows for accurate and efficient modeling of catalytic dynamics in complex systems like transition metal catalysts, which are often intractable with single-reference methods [67].
The CASSCF method is a powerful and sophisticated extension of the SCF paradigm, specifically designed to handle the challenges of strong electron correlation. Its success hinges on a careful and insightful selection of the active space. While CASSCF itself provides a qualitatively correct description, it serves as the essential foundation for more accurate multireference treatments of dynamic correlation, such as CASPT2 and MC-PDFT. Ongoing developments, including automated active space selection and protocols like WASP for machine learning applications, are pushing the boundaries of CASSCF, enabling its application to increasingly complex problems in catalysis and materials science.
Solving SCF Convergence Problems: Diagnostic and Remediation Strategies
The Self-Consistent Field (SCF) method represents the fundamental computational algorithm for solving electronic structure problems within Hartree-Fock and Density Functional Theory (DFT). This iterative procedure cycles between computing the electron density from a set of orbitals and generating new orbitals from that density until self-consistency is achieved [69]. Despite its widespread implementation, the SCF process does not always converge successfully. Convergence failures manifest in distinct patterns—oscillation, divergence, and stagnation—each indicating specific underlying physical or numerical issues within the quantum chemical calculation [70]. Understanding these patterns is crucial for researchers and drug development professionals who rely on computational chemistry for molecular modeling and property prediction, as these failures can halt critical calculations in pharmaceutical development pipelines.
The SCF iteration is inherently a nonlinear process, mathematically described by equations of the form ( x = f(x) ) [70]. This nonlinearity can lead to various behaviors during iteration, including clean convergence, oscillation between values, or seemingly random output. The convergence behavior is intimately connected to the electronic properties of the system under study and the numerical parameters governing the iterative process. This guide provides a comprehensive technical framework for identifying, diagnosing, and remediating the three primary SCF convergence failure patterns within the broader context of developing robust iterative processes for computational drug discovery applications.
Theoretical Framework: The SCF as a Nonlinear System
From a mathematical perspective, the SCF procedure constitutes a fixed-point iteration problem. The work of Young draws direct parallels between SCF convergence behaviors and chaos theory, where nonlinear systems can exhibit multiple stable states, oscillatory patterns between states, or even chaotic output depending on initial conditions and system parameters [70]. The local convergence of the SCF iteration can be analyzed through the spectral radius of the Jacobian of the fixed-point map, with convergence guaranteed only when this spectral radius is less than unity [71].
The density matrix provides a particularly insightful state representation for analyzing SCF convergence. Recent analytical work has established that convergence behavior is fundamentally governed by the eigenspectrum of the underlying quantum chemical system, particularly the energy gaps between occupied and unoccupied states [71]. Upper bounds on the convergence factor can be expressed in terms of these higher gaps, providing a theoretical explanation for why systems with small HOMO-LUMO gaps typically present greater convergence challenges.
Table 1: Fundamental SCF Convergence Theorems
| Mathematical Quantity | Impact on Convergence | Theoretical Bound |
|---|---|---|
| Spectral Radius of Jacobian | Determines local convergence | Must be <1 for convergence |
| HOMO-LUMO Gap | Primary determinant of convergence rate | Inverse relationship with convergence factor |
| Higher Energy Gaps (δ₂, δ₃, ...) | Secondary impact on convergence | Incorporated in modern convergence bounds |
Oscillation Failure Pattern
Identification and Characteristics
The oscillation failure pattern manifests as continuous cycling between two or more distinct electronic states without settling on a consistent solution. This pattern typically presents with energy oscillations having amplitudes ranging from (10^{-4}) to 1 Hartree [72]. In the context of chaos theory, these are known as "Lorenz attractor systems" or period-doubling scenarios where values oscillate between 2, 4, or other powers of 2 states [70]. Oscillation patterns can often be visualized by plotting the total energy or density matrix elements against iteration number, revealing clear periodic behavior rather than monotonic convergence.
Physical and Numerical Causes
The primary physical origin of oscillatory behavior is a small HOMO-LUMO gap, which enables repetitive changes in frontier orbital occupation numbers [72]. In such scenarios, an orbital that is occupied in one iteration may become unoccupied in the next, followed by a reversal in subsequent iterations. This electronic instability creates a feedback loop where the density matrix oscillates between different configurations. A second physical mechanism is charge sloshing, where the electron density oscillates between different spatial regions of the molecule, particularly problematic in systems with high polarizability [72]. From a numerical perspective, oscillation can result from overly aggressive DIIS acceleration or insufficient damping in the density matrix updates [70].
Diagnostic Protocols
To confirm oscillatory failure, researchers should:
- Plot total energy versus SCF iteration number to visualize the oscillation pattern
- Examine the orbital occupation numbers printed at the end of the SCF calculation for obvious occupation pattern errors
- Check the density matrix change between iterations for alternating patterns
- Calculate the HOMO-LUMO gap from the current orbital energies
Table 2: Oscillation Pattern Diagnostics and Solutions
| Diagnostic Check | Observation Indicating Oscillation | Recommended Solution |
|---|---|---|
| Energy vs. Iteration Plot | Clear periodic variation | Increase damping, use level shifting |
| Orbital Occupation Numbers | Alternating between iterations | Employ fractional occupation smearing |
| HOMO-LUMO Gap | Very small (<0.1 eV) | Use level shifting, adjust convergence algorithm |
| Density Matrix Changes | Large, alternating elements | Reduce DIIS space, conservative mixing |
Divergence Failure Pattern
Identification and Characteristics
Divergent SCF calculations display progressively increasing energy fluctuations with each iteration, ultimately leading to numerical overflow or termination. Unlike oscillation, which shows bounded variation, divergence exhibits unbounded growth in the energy error, often with error magnitudes exceeding 1 Hartree [72]. The electronic structure becomes increasingly unphysical with each iteration, typically accompanied by qualitatively incorrect orbital occupation patterns.
Physical and Numerical Causes
Divergence frequently stems from fundamentally flawed initial conditions, such as improper molecular geometry (e.g., atoms too close together), incorrect total charge, or unrealistic spin multiplicity [72] [73]. Geometries with artificially high symmetry that doesn't correspond to the true electronic state symmetry can also trigger divergence [72]. Numerical causes include near-linear dependence in the basis set or inadequate integration grids that create numerical noise amplified through successive iterations [72]. In open-shell systems, particularly those containing transition metals, an incorrect initial guess for the density matrix can initiate a divergent path [70].
Diagnostic Protocols
For divergent systems, researchers should:
- Verify molecular geometry合理性 (bond lengths, angles) and ensure correct units (angstroms vs. bohrs)
- Confirm the total charge and spin multiplicity match the chemical system
- Check for basis set linear dependence through overlap matrix conditioning
- Examine the initial density matrix guess for physical reasonableness
- Test with a minimal basis set to identify if the problem is basis-set related
Stagnation Failure Pattern
Identification and Characteristics
Stagnation presents as minimal progress toward convergence despite continued iteration, with energy changes remaining virtually unchanged (often below (10^{-4}) Hartree) but failing to meet the convergence threshold [72]. The calculation appears to "plateau" far from the true solution, continuing to run without meaningful improvement. This pattern can be particularly problematic as it may not trigger automatic termination, wasting computational resources.
Physical and Numerical Causes
Stagnation often results from insufficient variational flexibility in the electronic structure method or basis set to properly describe the system [73]. It can also occur when the SCF procedure is trapped in a shallow local minimum on the energy landscape, unable to make progress toward the global minimum. Numerical grid deficiencies that introduce consistent small errors or overly conservative convergence acceleration parameters can also cause stagnation [72]. In some cases, the problem arises from incorrect handling of symmetry constraints that conflict with the true electronic state [73].
Diagnostic Protocols
To address stagnation, researchers should:
- Monitor the rate of energy change over hundreds of iterations to confirm true stagnation
- Check for numerical noise in the integration grid or Hamiltonian construction
- Verify that molecular symmetry settings match the expected electronic state symmetry
- Test with different initial guesses to escape shallow local minima
- Examine the gradient norm to determine if the solution is genuinely stationary
Experimental Protocols for Convergence Remediation
Systematic Troubleshooting Methodology
When confronted with SCF convergence failures, researchers should implement a systematic troubleshooting protocol beginning with the most common and easily addressable issues. The first step involves verifying the fundamental calculation setup: confirming appropriate molecular geometry (including correct units), proper charge and multiplicity assignments, and physicality of the initial structure [7] [73]. For drug development applications, particular attention should be paid to metalloprotein active sites and unusual coordination environments that may challenge standard protocols.
The second tier involves modifying the SCF convergence acceleration parameters. Based on the observed failure pattern, different strategies are recommended. For oscillatory cases, increasing the damping factor (reducing the mixing parameter to 0.015-0.05) and expanding the DIIS subspace (N=15-25) can stabilize convergence [7]. For divergent cases, starting with a better initial guess—often from a lower-level calculation or a slightly different molecular geometry—is frequently effective [70]. For stagnant calculations, more aggressive convergence accelerators (LIST methods, ADIIS) or disabling symmetry can provide the necessary push toward convergence [69] [7].
Advanced Technical Interventions
When standard approaches fail, advanced interventions may be necessary. Level shifting artificially raises the energies of virtual orbitals, preventing unwanted occupation changes that drive oscillation [70]. This technique is particularly valuable for systems with small HOMO-LUMO gaps but must be used cautiously as it can affect properties involving virtual orbitals. Fractional orbital occupation (electron smearing) distributes electrons across near-degenerate orbitals, effectively increasing the HOMO-LUMO gap and damping oscillation [7]. For persistently problematic systems, quadratic convergence (QC) methods guarantee convergence at the expense of significantly increased computational cost per iteration [70].
SCF Failure Diagnosis
SCF Acceleration Methods and Parameter Optimization
Modern Acceleration Algorithms
Contemporary quantum chemistry packages offer multiple SCF acceleration algorithms beyond the traditional Pulay DIIS (Direct Inversion in the Iterative Subspace). The ADIIS (Augmented DIIS) method combines traditional DIIS with an energy-based interpolation approach, often providing superior performance for difficult systems [69]. The LIST (LInear-expansion Shooting Technique) family of methods represents another class of accelerators that can be effective where DIIS fails [69]. For particularly challenging cases, the MESA (Modular Eigensolver with Selection of Accelerators) method dynamically combines multiple acceleration techniques (ADIIS, fDIIS, LISTb, LISTf, LISTi, and SDIIS) to achieve convergence [69].
The Augmented Roothaan-Hall (ARH) method takes a different approach by directly minimizing the total energy as a function of the density matrix using a preconditioned conjugate-gradient method with a trust-radius approach [7]. While computationally more expensive per iteration, ARH can converge systems that prove intractable for conventional methods. Recent comparative studies indicate that different acceleration methods show varying effectiveness across chemical system types, suggesting that researchers should maintain flexibility in their approach to convergence acceleration [7].
Parameter Optimization Strategies
Optimal SCF parameter selection depends strongly on the specific failure pattern and chemical system. For oscillatory systems, reduced mixing parameters (0.01-0.05) combined with extended DIIS subspaces (N=15-25) and delayed DIIS onset (Cyc=20-30) typically improve stability [7]. A specific parameter set recommended for difficult systems includes DIIS N=25, DIIS Cyc=30, Mixing=0.015, and Mixing1=0.09 [7]. For systems suffering from stagnation, more aggressive mixing (0.3-0.5) and earlier DIIS activation (Cyc=2-3) may be beneficial.
Table 3: SCF Acceleration Methods and Applications
| Acceleration Method | Algorithm Type | Best For Failure Pattern | Key Parameters |
|---|---|---|---|
| Pulay DIIS (SDIIS) | Fock matrix extrapolation | Mild oscillation, Stagnation | DIIS N, Mixing |
| ADIIS | Energy interpolation + DIIS | Divergence, Strong oscillation | THRESH1, THRESH2 |
| LIST Methods | Linear expansion shooting | Stagnation, Oscillation | DIIS N |
| MESA | Hybrid multiple methods | All patterns, difficult cases | Component selection |
| Quadratic Convergence | Direct minimization | Intractable cases | Trust radius |
The Scientist's Toolkit: Research Reagent Solutions
Computational Reagents for SCF Convergence
Table 4: Essential Computational Reagents for SCF Convergence
| Reagent Solution | Function | Application Context |
|---|---|---|
| Level Shifting (VShift) | Artificially raises virtual orbital energies | Small HOMO-LUMO gaps, oscillation |
| Electron Smearing | Fractional orbital occupations | Metallic systems, near-degeneracy |
| DIIS Subspace Expansion | Increases extrapolation history | Oscillation, stagnation |
| Damping (Mixing) | Controls Fock matrix updates | Oscillation, divergence |
| Quadratic Convergence | Guaranteed convergence algorithm | Intractable cases |
| Basis Set Modification | Alters variational flexibility | Divergence from linear dependence |
| Symmetry Disabling | Removes constraint conflicts | Stagnation, incorrect state |
Implementation Protocols
For level shifting, typical values range from 0.1-0.5 Hartree for the virtual orbital energy increase. Most implementations allow automatic deactivation once the SCF error drops below a specified threshold (e.g., 0.01) to prevent affecting final results [69]. For electron smearing, the electronic temperature parameter should be kept as low as possible (0.001-0.01 Hartree) while still achieving convergence, with multiple restarts using successively smaller values to minimize impact on the final energy [7].
The DIIS subspace size significantly affects convergence stability. While default values typically range from 6-10, expanding to 15-25 vectors can stabilize oscillatory systems, though caution is warranted as excessively large subspaces can sometimes break convergence in small systems [69] [7]. Damping parameters control the fraction of the new Fock matrix included in each iteration, with lower values (0.01-0.05) providing greater stability for problematic cases, while higher values (0.3-0.5) can accelerate slow but stable convergence [7].
The systematic identification and remediation of SCF convergence failure patterns—oscillation, divergence, and stagnation—represents an essential competency for computational researchers in drug development and molecular sciences. Each pattern serves as a diagnostic signal pointing to specific underlying physical or numerical issues within the quantum chemical calculation. Through methodical application of the protocols outlined in this technical guide, researchers can significantly enhance the robustness and success rate of their electronic structure calculations, accelerating the discovery process in pharmaceutical applications and materials design. The continued development of SCF convergence methodology, particularly through the lens of nonlinear systems analysis and advanced acceleration algorithms, promises further improvements in the reliability of computational chemistry for complex biological and therapeutic systems.
The self-consistent field (SCF) method is the foundational algorithm for solving electronic structure problems within Hartree-Fock and density functional theory (DFT). It is an iterative procedure for finding a converged electronic state where the generating orbitals are consistent with the Fock matrix they generate [1] [13]. The central challenge is that the Fock matrix itself depends on the electron density, creating a nonlinear problem that must be solved iteratively [13]. The choice of the initial guess for the molecular orbitals or electron density is the most critical factor determining the success and speed of this process. A poor initial guess can lead to slow convergence, convergence to an incorrect electronic state, or complete SCF failure, particularly for challenging systems such as those with open-shell configurations, small HOMO-LUMO gaps, or containing heavy elements [74] [7].
This guide frames initial guess strategies within the broader context of optimizing the entire SCF iterative process. We explore the two advanced techniques highlighted in the title: the use of restart files from previous calculations and the application of external model potentials. These methods move beyond simple built-in guesses, offering researchers powerful tools to tackle increasingly complex systems in fields like drug development, where molecules often feature transition metals and complex electronic structures.
Core Theory: Initial Guess Types and Their Underlying Principles
The primary goal of an initial guess is to provide a starting point for the SCF cycle that is sufficiently close to the final, converged solution. The fundamental equation solved in each SCF iteration is the Fock matrix equation, F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital energies [1]. The following diagram illustrates the logical relationship between the different classes of initial guesses and their pathways to generating a density matrix for the first SCF iteration.
Table 1: Comparison of Common Initial Guess Methods
| Guess Type | Theoretical Basis | Typical Keywords | Advantages | Disadvantages |
|---|---|---|---|---|
| Core Hamiltonian | Diagonalizes the one-electron core Hamiltonian matrix, ignoring electron-electron repulsion [74] [41]. | Guess=HCore (ORCA) [74], SCF_GUESS=CORE (Q-Chem) [41], init_guess='1e' (PySCF) [1] |
Simple and fast to compute. | Often a poor guess for molecular systems; produces overly compact orbitals and incorrect atomic shell structure [41]. |
| SAD & PAtom | Superposition of Atomic Densities (SAD) or precomputed atomic SCF orbitals [74] [41]. | Guess=PAtom (ORCA) [74], SCF_GUESS=SAD (Q-Chem) [41], init_guess='atom' (PySCF) [1] |
Generally robust; reflects molecular shape; good for heavy elements [74] [13]. | SAD density is non-idempotent, requiring at least two SCF iterations [41]. |
| PModel / SAP | Superposition of Atomic Potentials (SAP) or model DFT potentials from neutral, spherical atoms [74] [41]. | Guess=PModel (ORCA) [74], SCF_GUESS=SAP (Q-Chem) [41], init_guess='vsap' (PySCF) [1] |
Excellent for heavy elements; correctly describes atomic shell structure [74] [41]. | More computationally expensive than simpler guesses [74]. |
| Hückel | Performs an extended Hückel calculation in a minimal basis (e.g., STO-3G) and projects orbitals onto the target basis [74] [42]. | Guess=Hueckel (ORCA) [74], Guess=Huckel (Gaussian) [42] |
Includes some molecular information. | Quality can be limited by the poor minimal basis set [74]. |
Methodology: A Practical Guide to Advanced Initial Guess Strategies
Using Restart Files and Projecting Molecular Orbitals
Using orbitals from a previously converged calculation is often the best possible initial guess, as it can be very close to the final solution. This is typically done by reading a restart file containing the molecular orbitals [74] [1].
Experimental Protocol: Restarting from a Previous Calculation
Generate a Restart File: In your initial calculation, ensure the program writes a checkpoint or restart file (e.g., a
.gbwfile in ORCA, a.chkfile in Gaussian/PySCF, or a.wfnfile in CP2K). This is often the default behavior.Read the Restart File: In the subsequent calculation, specify the restart file using the appropriate input commands.
Handle Basis Set/Geometry Changes (Projection): A powerful feature of the restart method is that the initial calculation and the new calculation do not need to have the same geometry or basis set. The program will automatically project the old orbitals onto the new basis set [74] [1]. ORCA provides two projection methods controlled by
GuessMode:GuessMode FMatrix: Simpler and faster. Defines an effective one-electron operator and diagonalizes it in the new basis [74].GuessMode CMatrix: More involved. Uses the theory of corresponding orbitals to fit each MO subspace (occupied, virtual) separately, which can be advantageous for restarting ROHF calculations [74].
Special Cases: For calculations where redundant components were removed from the basis set, ORCA requires the use of
! rescue moread(withoutnoiter) to avoid incorrect results [74]. The workflow for implementing a restart calculation is summarized in the diagram below.
Exploiting External Potentials and Model Potentials
The PModel guess in ORCA and the Superposition of Atomic Potentials (SAP) guess in Q-Chem and PySCF represent a superior class of initial guesses that leverage external potential information [74] [41] [1].
Experimental Protocol: Employing a Model Potential Guess
Principle: These methods construct a guess by building and diagonalizing a Kohn-Sham matrix with an electron density composed of a superposition of predetermined, spherically averaged neutral atom densities (
PModel) or potentials (SAP) [74] [41].Implementation:
- ORCA: Use the
Guess PModeldirective within the%scfblock or the simple!PModelkeyword in the input line [74]. - Q-Chem: Set
SCF_GUESS = SAPin the$remsection. The numerical grid for constructing the potential can be controlled with theGUESS_GRIDvariable [41]. - PySCF: For DFT calculations, use
mf.init_guess = 'vsap'[1].
- ORCA: Use the
When to Use: This is the recommended method for systems containing heavy elements, as the model densities/potentials are available for most of the periodic table. It is valid for both Hartree-Fock and DFT methods and typically requires less computational time than a single SCF iteration [74].
The Scientist's Toolkit: Key Computational Reagents
Table 2: Essential "Research Reagents" for SCF Initial Guess Optimization
| Item / Keyword | Software Package | Function |
|---|---|---|
| MORead / moread | ORCA [74] | Instructs the SCF solver to read the initial guess molecular orbitals from a specified restart file. |
| chkfile | PySCF [1] | The file path to a checkpoint file from a previous calculation, used to read the initial guess. |
| PModel / SAP Guess | ORCA, Q-Chem, PySCF [74] [41] [1] | Generates an initial guess from a superposition of atomic densities or potentials, often the most robust choice. |
| AutoStart / !NoAutoStart | ORCA [74] | A feature that automatically uses an existing .gbw file of the same name as the initial guess. Can be disabled if not desired. |
| GuessMode | ORCA [74] | Controls the method (FMatrix or CMatrix) for projecting orbitals from a restart file onto a new basis set or geometry. |
| DIIS | All major packages [7] [1] | (Direct Inversion in the Iterative Subspace) The standard algorithm for accelerating SCF convergence by extrapolating Fock matrices. |
| Level Shift | PySCF, ADF [7] [1] | A technique to stabilize convergence by artificially increasing the energy of virtual orbitals, widening the HOMO-LUMO gap. |
| Damping | PySCF, ADF [7] [1] | Mixes a fraction of the previous Fock matrix with the new one to slow down and stabilize the SCF evolution. |
Troubleshooting and Convergence Acceleration
Even with a good initial guess, some systems remain challenging to converge. Here are key strategies to employ when standard procedures fail.
Verify Physical Assumptions: Before adjusting technical parameters, ensure the calculation setup is physically sound. Confirm the molecular geometry is reasonable, the spin multiplicity is correct for the expected electronic state, and the system charge is accurate [7] [73]. An improperly described electronic structure is a common root cause of convergence failure [7].
Tune SCF Convergence Accelerators:
- DIIS Parameters: Most codes use DIIS by default. For difficult cases, increasing the number of DIIS vectors (e.g., from 10 to 25) and applying DIIS after more initial equilibration cycles can enhance stability [7].
- Damping and Mixing: Using a lower mixing parameter (e.g., 0.015 instead of 0.2) slows down the convergence but can prevent oscillations in problematic systems [7]. In PySCF, this is controlled with
mf.damp[1]. - Level Shifting: Artificially raising the energy of virtual orbitals can quench oscillatory behavior in systems with small HOMO-LUMO gaps [7] [1].
Advanced Techniques:
- Fractional Occupation / Smearing: Assigning fractional occupations to orbitals near the Fermi level can help converge metallic systems or those with many near-degenerate states [7] [1].
- Second-Order SCF (SOSCF): Methods like Newton's method can achieve quadratic convergence and are invoked in PySCF via
.newton()[1]. For grand-canonical SCF, manifold optimization techniques have shown superior efficiency over standard DIIS [75]. - Alternate Algorithms: If DIIS fails, other algorithms like the Augmented Roothaan-Hall (ARH) method, which directly minimizes the total energy, can be viable alternatives [7].
Optimizing the initial guess is a critical step in ensuring the robustness and efficiency of SCF calculations. Within the broader framework of SCF iterative process guidance, two powerful strategies stand out: the use of restart files from previous calculations, which provides a near-optimal starting point, and the application of external model potentials like PModel and SAP, which offer a robust, ab initio starting point even for complex systems with heavy elements. Mastering these techniques, along with knowing how to troubleshoot convergence failures, is indispensable for researchers and drug development professionals pushing the boundaries of electronic structure theory. By strategically selecting and applying these methods, computational scientists can significantly expand the range of systems they can study reliably and efficiently.
The self-consistent field (SCF) method is a cornerstone computational technique in quantum chemistry, employed in both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (KS-DFT) to determine the electronic structure of atoms, molecules, and materials. The SCF procedure involves an iterative process where an initial guess of the molecular orbitals is progressively refined until the solution becomes self-consistent, meaning the output orbitals and electron density remain unchanged between successive iterations. This process is governed by the Roothaan-Hall equation, F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital energies [1].
Despite its fundamental importance, the SCF process often encounters significant convergence challenges, particularly for systems with complex electronic structures such as open-shell transition metal complexes, molecules with small highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) gaps, or cases with degenerate or near-degenerate states. These challenges manifest as oscillatory behavior, slow convergence, or complete divergence of the iterative process. To address these issues, computational chemists have developed several convergence acceleration techniques, among which damping, level shifting, and fractional occupation methods have proven particularly valuable. This guide provides an in-depth technical examination of these three core techniques, framing them within the broader context of robust SCF methodology for research and drug development applications.
The SCF Iterative Process: A Foundation for Understanding Convergence Issues
The standard SCF iterative procedure follows a well-defined cycle, which can be summarized in the following steps:
- Initialization: Generate an initial guess for the density matrix or molecular orbitals using methods such as superposition of atomic densities ('minao' or 'atom' guesses), core Hamiltonian diagonalization ('1e' guess), or reading from a previous calculation ('chk' guess) [1].
- Fock Matrix Construction: Build the Fock matrix using the current density matrix. This involves calculating the one-electron integrals (kinetic energy and nuclear attraction) and two-electron integrals (Coulomb and exchange terms).
- Fock Matrix Diagonalization: Solve the Roothaan-Hall equation F C = S C E to obtain an updated set of molecular orbitals and their energies.
- Density Matrix Update: Construct a new density matrix from the occupied molecular orbitals according to the Aufbau principle.
- Convergence Check: Assess whether the calculation has converged based on changes in energy, density matrix, or other criteria. If convergence is achieved, the calculation terminates; otherwise, the cycle repeats from step 2.
The convergence of this process can be assessed through multiple criteria, with typical thresholds as shown in Table 1.
Table 1: Standard SCF Convergence Criteria in Quantum Chemistry Codes
| Criterion | Description | TightSCF Values [76] | StrongSCF Values [76] |
|---|---|---|---|
| TolE | Energy change between cycles | 1e-8 | 3e-7 |
| TolRMSP | Root-mean-square density change | 5e-9 | 1e-7 |
| TolMaxP | Maximum density change | 1e-7 | 3e-6 |
| TolErr | DIIS error vector norm | 5e-7 | 3e-6 |
| TolG | Orbital gradient convergence | 1e-5 | 2e-5 |
The following diagram illustrates the core SCF process and points where convergence issues typically arise:
Diagram 1: The SCF iterative process with common convergence challenges.
Damping Techniques for SCF Stabilization
Theoretical Foundation of Damping
Damping is one of the oldest and simplest SCF acceleration techniques, with origins dating back to Hartree's early work on atomic structures [77]. The fundamental principle behind damping is the linear mixing of the current density (or Fock) matrix with that from the previous iteration to reduce large oscillations that can occur, particularly in the early stages of the SCF process. The mathematical formulation of density matrix damping is given by:
Pndamped = (1 - α) Pn + α Pn-1
where Pn is the density matrix from the current iteration, Pn-1 is the density matrix from the previous iteration, and α is the damping factor (mixing parameter) with values between 0 and 1 [77]. A value of α = 0 corresponds to no damping, while higher values increase the mixing with the previous density matrix.
Practical Implementation and Parameters
In practical implementations, damping is often combined with more advanced acceleration techniques like DIIS. For instance, the DP_DIIS algorithm applies damping for a limited number of initial cycles before switching to standard DIIS extrapolation [77]. Key parameters controlling damping behavior include:
- NDAMP: Determines the mixing coefficient (α = NDAMP/100), typically set between 50-75 [77].
- MAXDPCYCLES: The maximum number of SCF iterations with damping before it is automatically turned off.
- THRESHDPSWITCH: The threshold for turning off damping based on the magnitude of density matrix changes.
Table 2: Damping Parameters and Their Effects in SCF Calculations
| Parameter | Typical Values | Effect on SCF Convergence | Recommended Usage |
|---|---|---|---|
| Damping Factor (α) | 0.5 - 0.75 | Reduces oscillations but may slow convergence | Increase for systems with strong SCF fluctuations |
| Damping Start Cycle | 0-2 | When damping begins in SCF procedure | Earlier start for problematic systems |
| Damping Duration | 3-20 cycles | How long damping remains active | Longer for persistently oscillating systems |
| Damping Termination | Based on density change | Automatic shutoff when SCF stabilizes | Prevents unnecessary slowdown |
Damping is particularly effective during the initial SCF iterations when the density matrix may change dramatically between cycles. However, if applied throughout the entire SCF process, it can unnecessarily slow convergence once the procedure has stabilized. Therefore, modern implementations typically employ damping only in the early stages or when specific instability thresholds are exceeded.
Level Shifting for Gap Enhancement
Principles of Level Shifting
Level shifting addresses SCF convergence problems by artificially increasing the energy gap between occupied and virtual orbitals. Systems with small HOMO-LUMO gaps are particularly prone to convergence difficulties because even small changes in the density matrix can cause significant mixing between occupied and virtual orbitals, leading to oscillations or divergence [1]. The level shifting technique modifies the Fock matrix according to:
Fμνshifted = Fμν + σ ∑i CμiCνi
where σ is the level shift parameter (typically positive, with common values between 0.1 and 1.0 Hartree), and the sum runs over all virtual orbitals [1]. This modification effectively raises the energy of virtual orbitals, increasing the HOMO-LUMO gap and reducing orbital mixing.
Implementation Considerations
The implementation of level shifting requires careful consideration of the shift magnitude. While larger values provide more stabilization, excessive shifting can dramatically slow convergence by making the orbital updates too conservative. Dynamic level shifting schemes that adjust the shift parameter based on SCF behavior have been developed to balance these competing concerns [1]. In the PySCF package, level shifting is controlled through the level_shift attribute, with examples provided in the documentation demonstrating its application to challenging systems [1].
Fractional Occupation Techniques
Theoretical Basis for Fractional Occupations
Fractional occupation number techniques address SCF convergence challenges by temporarily relaxing the strict Aufbau principle that assigns integer occupations (0 or 2 for closed-shell systems) to molecular orbitals. Instead, these methods allow for continuous occupation numbers between 0 and 2, particularly for orbitals near the Fermi level [1]. This approach is physically justified for systems with degenerate or near-degenerate states at the Fermi level and can significantly improve convergence by smoothing the energy landscape.
The mathematical formulation involves modifying the density matrix construction:
Pμν = ∑i fi CμiCνi
where fi are now fractional occupation numbers determined by a chosen distribution function, such as the Fermi-Dirac distribution:
fi(ε) = 1 / [1 + exp((εi - εF)/kT)]
where εF is the Fermi energy, k is Boltzmann's constant, and T is an effective electronic temperature [1].
Smearing Methods and Implementation
Fractional occupation techniques are often implemented through smearing methods, which assign partial occupations according to a temperature-dependent function. Common smearing approaches include:
- Fermi-Dirac smearing: Uses the Fermi-Dirac distribution for occupations
- Gaussian smearing: Applies Gaussian broadening to occupation numbers
- Methfessel-Paxton smearing: Uses a series expansion approach for faster convergence
These methods are particularly valuable for metallic systems, molecules with significant static correlation, or cases where symmetry breaking causes convergence difficulties. The PySCF package provides examples of fractional occupancy implementation, demonstrating improved convergence for challenging systems [1].
Advanced Combined Methods and Implementation Protocols
DIIS-Based Acceleration Methods
The Direct Inversion in the Iterative Subspace (DIIS) method represents a more sophisticated approach to SCF acceleration. DIIS extrapolates the Fock matrix by minimizing the norm of the commutator [F,PS] where P is the density matrix and S is the overlap matrix [1] [46]. Several DIIS variants have been developed:
- Traditional DIIS: Minimizes the error vector derived from the commutator of Fock and density matrices [46]
- EDIIS (Energy DIIS): Minimizes a quadratic energy function to obtain linear coefficients [46]
- ADIIS (Augmented DIIS): Uses the augmented Roothaan-Hall energy function for coefficient determination [46]
Research has demonstrated that combined approaches such as "ADIIS+DIIS" can be highly reliable and efficient for accelerating SCF convergence [46]. The OpenOrbitalOptimizer library implements these standard algorithms (DIIS, EDIIS, ADIIS, ODA) as reusable components for quantum chemistry codes [78].
Experimental Protocol for Challenging Systems
For researchers dealing with particularly challenging SCF convergence problems (e.g., open-shell transition metal complexes), the following systematic protocol is recommended:
- Begin with an improved initial guess using 'atom' or 'huckel' guesses rather than the default [1]
- Apply moderate damping (α = 0.5-0.7) for the first 5-10 iterations [77]
- Implement a small level shift (0.1-0.3 Hartree) if oscillations persist [1]
- Introduce fractional occupations with moderate smearing (0.001-0.01 Hartree) if the system has small HOMO-LUMO gaps [1]
- Apply DIIS acceleration once the SCF has stabilized, using ADIIS+DIIS for difficult cases [46]
- Progressively remove accelerators (first fractional occupations, then level shifting, then damping) as convergence approaches
- Perform stability analysis on the converged solution to ensure it represents a true minimum [1]
The relationship between these techniques and their application sequence can be visualized as follows:
Diagram 2: Sequential application of convergence accelerators for challenging systems.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for SCF Convergence Research
| Tool/Resource | Type | Function in SCF Convergence | Implementation Examples |
|---|---|---|---|
| OpenOrbitalOptimizer | Software Library | Provides reusable implementation of DIIS, EDIIS, ADIIS, ODA algorithms | Easy interfacing with legacy quantum chemistry codes [78] |
| PySCF | Quantum Chemistry Package | Implements damping, level shifting, fractional occupations, DIIS variants | Python-based, modular platform for SCF experimentation [1] |
| ORCA | Quantum Chemistry Program | Offers comprehensive convergence controls with predefined criteria sets | Convergence keywords (TightSCF, StrongSCF) with associated parameters [76] |
| Q-Chem | Quantum Chemistry Software | Implements combined damping-DIIS algorithms (DPDIIS, DPGDM) | SCFALGORITHM = DPDIIS with NDAMP, MAXDPCYCLES controls [77] |
Damping, level shifting, and fractional occupation techniques represent essential components of the modern computational chemist's toolkit for addressing SCF convergence challenges. While each method operates through distinct mechanisms—damping through linear mixing of density matrices, level shifting through artificial gap enhancement, and fractional occupations through occupation smoothing—they share the common goal of stabilizing the SCF process and facilitating convergence for electronically challenging systems.
The effectiveness of these techniques is maximized when they are applied strategically, either individually or in combination, with careful attention to system-specific requirements and parameter optimization. For researchers in drug development and materials science, where computational efficiency and reliability are paramount, mastering these convergence accelerators can significantly enhance productivity and enable the study of increasingly complex molecular systems.
As quantum chemistry software continues to evolve, we anticipate further refinement of these established techniques alongside the development of novel approaches that will expand the range of tractable electronic structure problems while improving computational efficiency.
The Self-Consistent Field (SCF) method is a cornerstone computational procedure in quantum chemistry for solving the electronic structure problem within the Hartree-Fock (HF) and Kohn-Sham Density Functional Theory (KS-DFT) frameworks [4] [20]. This iterative method seeks to find a set of molecular orbitals where the field generated by the electrons is consistent with their own spatial distribution. The fundamental SCF equation, often expressed as the Roothaan-Hall equation FC = SCE, constitutes a nonlinear eigenvalue problem that must be solved iteratively [1] [20]. During this process, the Fock or Kohn-Sham matrix (F) is built from the current density matrix (P), which is then used to compute a new density matrix, creating a cyclic dependency that must be resolved until self-consistency is achieved [4].
A significant challenge in SCF calculations is that this iterative process does not always converge smoothly. Systems with small HOMO-LUMO gaps, such as metals or open-shell transition metal complexes, often exhibit oscillatory or divergent behavior [1] [79]. To mitigate these issues and accelerate convergence, sophisticated algorithmic techniques are essential. The Direct Inversion in the Iterative Subspace (DIIS) method, developed by Peter Pulay, has become the most widely used acceleration technique [80] [1]. This guide provides an in-depth examination of DIIS parameter tuning, mixing strategies, and cycle control mechanisms to help researchers optimize SCF convergence, particularly for challenging systems encountered in drug development and materials science.
Core Concepts: DIIS, Mixing, and Cycle Control
The DIIS Method
The DIIS method accelerates SCF convergence by extrapolating a new Fock matrix as a linear combination of Fock matrices from previous iterations. The core idea relies on the property that at self-consistency, the density and Fock matrices must commute, satisfying the condition SPS - FPS = 0, where S is the overlap matrix [80]. Before convergence, this commutator defines an error vector, eᵢ = SPᵢFᵢ - FᵢPᵢS, which is non-zero [80]. DIIS determines optimal coefficients for combining previous Fock matrices by minimizing the norm of the combined error vectors subject to a normalization constraint (∑cₖ = 1), leading to a system of linear equations [80]:
Mixing Techniques
Mixing strategies combine information from previous iterations to generate a better input for the next cycle. While DIIS mixes Fock matrices, alternative approaches mix the density matrix or Hamiltonian directly [69] [79]. Linear mixing applies simple damping: Xnew = (1-α)Xold + αX_calculated, where X represents the mixed quantity and α is the mixing factor [79]. More advanced methods like Pulay (DIIS) mixing and Broyden mixing utilize historical data to construct better updates [79]. The optimal mixing strategy depends on the system; metallic systems often benefit from Broyden or Kerker mixing, while molecular systems typically perform well with Pulay methods [79].
Cycle Control Parameters
Effective SCF cycle management involves strategic phase transitions during the convergence process:
- Damping Phase: Initial iterations often employ simple damping to stabilize early oscillations
- DIIS Activation: Switching to DIIS after initial stabilization prevents the introduction of large errors early in the process [1]
- Subspace Management: Limiting the DIIS subspace size prevents numerical instability and memory bloat [80]
- Convergence Criteria: Setting appropriate tolerances for energy, density, and DIIS error ensures reliable convergence without unnecessary iterations [9]
Quantitative Parameter Analysis Across Quantum Chemistry Codes
Table 1: Default DIIS and Mixing Parameters in Various Quantum Chemistry Codes
| Code | Default DIIS Subspace Size | Default Mixing Factor | DIIS Variants Supported | Key Control Parameters |
|---|---|---|---|---|
| Q-Chem | 15 [80] | Not specified | Standard DIIS, ADIIS [80] | DIIS_SUBSPACE_SIZE, DIIS_ERR_RMS [80] |
| ADF | 10 [69] | 0.2 [69] | SDIIS, ADIIS, LIST methods [69] | DIIS N, DIIS OK, DIIS Cyc [69] |
| PySCF | Not specified | Damping: 0.5 [1] | EDIIS, ADIIS [1] | damp, diis_start_cycle, level_shift [1] |
| SIESTA | History: 2 [79] | Weight: 0.25 [79] | Pulay, Broyden [79] | SCF.Mixer.History, SCF.Mixer.Weight [79] |
| BAND | Not specified | 0.075 [50] | DIIS, MultiSecant, MultiStepper [50] | SCF Method, SCF Mixing [50] |
Table 2: SCF Convergence Tolerance Presets in ORCA [9]
| Convergence Level | Energy Tolerance (TolE) | Max Density Tolerance (TolMaxP) | DIIS Error Tolerance (TolErr) | Typical Use Case |
|---|---|---|---|---|
| Loose | 1e-5 | 1e-3 | 5e-4 | Preliminary calculations |
| Medium | 1e-6 | 1e-5 | 1e-5 | Standard single-point |
| Strong | 3e-7 | 3e-6 | 3e-6 | Geometry optimization |
| Tight | 1e-8 | 1e-7 | 5e-7 | Transition metal complexes |
| VeryTight | 1e-9 | 1e-8 | 1e-8 | Frequency calculations |
Table 3: Recommended DIIS Subspace Sizes for Different System Types
| System Characteristic | Recommended DIIS Subspace Size | Rationale |
|---|---|---|
| Small molecules (<10 atoms) | 5-10 [69] | Prevents overfitting and numerical issues |
| Medium systems (10-50 atoms) | 10-15 [80] | Balances history and computational cost |
| Large systems (>50 atoms) | 15-20 | Provides sufficient history for complex systems |
| Metallic systems | 15-20 [79] | Addresses charge sloshing with more history |
| Difficult convergence cases | 12-20 [69] | Additional history helps overcome oscillations |
Experimental Protocols for Parameter Optimization
Systematic DIIS Subspace Size Optimization
Objective: Determine the optimal DIIS subspace size for efficient SCF convergence.
Materials: Quantum chemistry software (Q-Chem, ADF, PySCF, ORCA), molecular system of interest, computational resources.
Methodology:
- Initial Setup: Begin with a standard initial guess ('minao' in PySCF or 'atom' in ADF) and fixed convergence criteria (e.g., Medium in ORCA) [69] [1]
- Baseline Calculation: Run SCF with the default DIIS subspace size (typically 10-15) to establish reference convergence behavior [80]
- Subspace Variation: Perform a series of calculations with DIIS subspace sizes ranging from 5 to 25 in increments of 2-5
- Data Collection: Record for each run:
- Total SCF iteration count
- Calculation time per iteration
- Convergence history (energy and density changes)
- Any numerical warnings or errors
- Analysis: Identify the subspace size that minimizes the product of iteration count and time per iteration
Expected Outcomes: Most molecular systems show optimal performance with subspace sizes of 8-15, while challenging systems may require 15-20 [80] [69]. Excessively large subspaces (>25) may cause numerical instability without improving convergence.
Mixed DIIS-Damping Protocol for Problematic Systems
Objective: Establish a robust SCF procedure for systems with persistent convergence difficulties.
Methodology:
- Initial Damping Phase:
- DIIS Activation:
- Switch to DIIS acceleration after the initial damping phase
- Begin with a moderately sized subspace (10-15)
- Convergence Monitoring:
- Track both energy and density matrix changes
- If oscillations occur, increase damping or implement level shifting (0.001-0.01 Hartree) [1]
- Fallback Strategy:
Validation: For transition metal complexes, this protocol typically reduces iteration count by 30-50% compared to standard DIIS while improving reliability.
Mixing Factor and History Optimization for Metallic Systems
Objective: Optimize Pulay or Broyden mixing parameters for metallic systems with delocalized electrons.
Methodology:
- Initial Assessment:
- Run calculations with default mixing parameters
- Observe convergence behavior, particularly "charge sloshing" patterns
- Parameter Screening:
- Test mixing weights from 0.05 to 0.5 in 0.05 increments
- Evaluate history depths from 2 to 10
- Compare Hamiltonian versus density matrix mixing [79]
- Performance Metrics:
- Primary: Number of SCF iterations to convergence
- Secondary: Stability of convergence (monotonic vs. oscillatory)
- Tertiary: Computational cost per iteration
- Optimal Parameter Selection:
- Metallic systems typically require smaller mixing weights (0.05-0.15) and larger history (7-10) [79]
- Molecular systems perform better with larger mixing weights (0.2-0.3) and moderate history (4-7)
Visualization of SCF Convergence Control Workflow
SCF Convergence Optimization Workflow: A decision pathway for systematic SCF convergence optimization, incorporating damping, DIIS acceleration, and troubleshooting strategies.
Table 4: Essential Computational Reagents for SCF Convergence Research
| Tool Category | Specific Implementation | Function in SCF Research | Availability |
|---|---|---|---|
| DIIS Variants | Standard Pulay DIIS [80] | Basic Fock matrix extrapolation | Q-Chem, PySCF, ADF |
| ADIIS (Augmented DIIS) [69] | Improved stability for difficult cases | ADF, Q-Chem | |
| EDIIS (Energy DIIS) [1] | Energy-based convergence | PySCF | |
| Mixing Algorithms | Linear Mixing [79] | Simple damping for initial cycles | SIESTA, BAND |
| Pulay Mixing [79] | Default efficient mixing | SIESTA, ADF | |
| Broyden Mixing [79] | Advanced mixing for metals | SIESTA | |
| Stabilization Methods | Level Shifting [1] | Increases HOMO-LUMO gap | PySCF, ADF |
| Fermi Smearing [1] | Fractional occupations | PySCF, ORCA | |
| SOSCF [1] | Second-order convergence | PySCF | |
| Analysis Tools | Stability Analysis [1] | Verify solution quality | PySCF, ORCA |
| Error Vector Monitoring [80] | DIIS performance assessment | Q-Chem, ADF |
Advanced DIIS Control Strategies
Adaptive DIIS Techniques
Modern implementations often employ adaptive DIIS strategies that automatically adjust parameters during the SCF procedure. The ADF code implements an adaptive approach where DIIS coefficients are weighted based on the current error magnitude [69]. In this scheme, ADIIS dominates at high errors (>0.01), while standard DIIS takes over as convergence approaches (error <0.0001), with a smooth transition between these regimes [69]. This hybrid approach combines the global convergence properties of ADIIS with the local efficiency of standard DIIS.
DIIS Subspace Management
Effective subspace management is crucial for maintaining numerical stability. The DIIS matrix can become ill-conditioned as calculations proceed, necessitating occasional resets [80]. Most codes implement safeguards when the DIIS coefficients become excessively large (e.g., >20), either switching to damping or removing the oldest vectors from the subspace [50]. For extended calculations, limiting the subspace size to 15-20 vectors prevents numerical problems while maintaining convergence efficiency [80].
System-Specific DIIS Recommendations
- Transition Metal Complexes: Use larger DIIS subspaces (15-20) with initial damping and potential level shifting [1] [9]
- Open-Shell Systems: Consider separate error vectors for alpha and beta spins to prevent false convergence [80]
- Metallic Systems: Implement smaller mixing factors (0.05-0.15) with Broyden or Kerker mixing as alternatives to DIIS [79]
- Large Biomolecular Systems: Use moderate subspace sizes (10-15) to balance memory usage and convergence performance
Strategic tuning of DIIS parameters, mixing factors, and cycle control mechanisms significantly enhances the reliability and efficiency of SCF calculations. The optimal configuration is highly system-dependent, requiring researchers to methodically explore parameter spaces using the protocols outlined herein. For drug development applications involving complex molecular systems, a mixed strategy combining initial damping with subsequent DIIS acceleration typically provides the most robust performance. As quantum chemistry continues to advance toward larger and more complex systems, mastering these SCF convergence techniques remains essential for producing accurate and computationally efficient results.
The self-consistent field (SCF) method serves as the fundamental algorithm for determining electronic structure configurations within both Hartree-Fock theory and Kohn-Sham density functional theory [7] [1]. This iterative procedure minimizes the total electronic energy subject to orbital orthogonality constraints, effectively describing electrons as independent particles interacting through a mean field [1]. Despite its widespread implementation across computational chemistry packages, SCF calculations frequently encounter convergence difficulties in specific chemical systems. These challenges most commonly arise in systems exhibiting very small HOMO-LUMO gaps, those containing d- and f-elements with localized open-shell configurations, and in transition state structures with dissociating bonds [7]. Additional problems may stem from non-physical calculation setups, including high-energy geometries or an inappropriate initial description of the electronic structure [7]. This technical guide examines the core challenges and solutions for these problematic systems within the broader context of SCF iterative process research.
Fundamental SCF Theory and the Convergence Problem
The SCF method derives from the variational solution of the Hartree-Fock or Kohn-Sham equations. The central equation takes the form:
F C = S C E
Here, F represents the Fock matrix, C is the matrix of molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital eigenenergies [1]. The Fock matrix itself is composed of several components: F = T + V + J + K, where T is the kinetic energy matrix, V is the external potential, J is the Coulomb matrix, and K is the exchange matrix [1]. Since the Fock matrix depends on the electron density through the Coulomb and exchange terms, which in turn depend on the molecular orbitals, the equation must be solved self-consistently through an iterative procedure.
The convergence problems in challenging systems often originate from the mathematical structure of the SCF equations. In systems with small HOMO-LUMO gaps, the energetic ordering of orbitals can switch during optimization, creating discontinuities [81]. For open-shell systems, the proper description of spin polarization and the potential for symmetry breaking introduce additional complexity [82]. In metallic systems with vanishing band gaps, the electron delocalization creates intrinsic instability in the density matrix updates [81].
Table 1: Common SCF Convergence Problems and Their Physical Origins
| System Type | Primary Challenge | Electronic Structure Manifestation |
|---|---|---|
| Small-Gap Systems | Near-degenerate frontier orbitals | Orbital ordering switches during optimization, causing discontinuities [81] |
| Open-Shell Configurations | Unpaired electrons and spin polarization | Improper spin coupling or symmetry breaking [7] [82] |
| Metallic Systems | Vanishing HOMO-LUMO gap | Excessive mixing between occupied and virtual orbitals [81] |
| Transition Metal Complexes | Localized d/f-orbitals | Multiple nearly degenerate electronic states [7] |
Technical Strategies for Challenging Systems
Initial Guess Improvement Techniques
The initial guess for the electron density matrix critically influences SCF convergence behavior. A poor initial guess can lead to oscillations or divergence in the iterative process. Several sophisticated methods exist for generating improved initial guesses:
- Superposition of Atomic Densities (
minao/atom): Projects minimal basis set atomic orbitals onto the target basis set, or employs spin-restricted atomic Hartree-Fock calculations with spherically averaged fractional occupations [1]. - Parameter-free Hückel Guess (
huckel): Utilizes on-the-fly atomic Hartree-Fock calculations to build a Hückel-type matrix that is diagonalized to obtain guess orbitals [1]. - Superposition of Atomic Potentials (
vsap): Applies pretabulated, fully numerical atomic potentials to build a guess potential on a DFT quadrature grid (DFT-only) [1]. - Restart from Checkpoint Files (
chk): Reads orbitals from previous calculations, which can be particularly effective when using a converged wavefunction from a similar chemical system or smaller basis set [1] [83].
For particularly difficult cases, such as the chromium atom in a septet state, a strategic approach involves first converging the calculation for the Cr⁶⁺ cation (which has a closed-shell configuration), and then using the resulting density matrix as an initial guess for the neutral atom calculation [1].
SCF Convergence Acceleration Algorithms
Several mathematical algorithms have been developed to accelerate and stabilize SCF convergence:
- DIIS (Direct Inversion in Iterative Subspace): The default method in many codes, DIIS extrapolates the Fock matrix by minimizing the norm of the commutator [F, PS] using Fock matrices from previous iterations [1]. Key parameters include:
- EDIIS/ADIIS: Enhanced DIIS variants that can provide improved convergence in difficult cases [1].
- MESA, LISTi, EDIIS: Alternative convergence acceleration methods available in the ADF package with differing performance characteristics across various chemical systems [7].
- SOSCF (Second-Order SCF): Implements a co-iterative augmented Hessian method to achieve quadratic convergence in orbital optimization, invoked in PySCF via the
.newton()decorator [1]. - ARH (Augmented Roothaan-Hall): Directly minimizes the total energy as a function of the density matrix using a preconditioned conjugate-gradient method with a trust-radius approach [7].
Table 2: SCF Acceleration Methods and Their Applications
| Method | Algorithmic Approach | Best For | Computational Cost |
|---|---|---|---|
| DIIS | Linear extrapolation of Fock matrix | General purpose systems | Low |
| EDIIS/ADIIS | Energy-bias and error minimization | Systems near convergence | Moderate |
| SOSCF | Second-order orbital optimization | Systems with strong coupling | High |
| ARH | Direct density matrix minimization | Problematic open-shell systems | Highest |
| MESA/LISTi | Alternative subspace methods | Metallic and small-gap systems | Variable |
System-Specific Convergence Protocols
Small HOMO-LUMO Gap and Metallic Systems
Systems with small or vanishing HOMO-LUMO gaps require specialized techniques to ensure convergence:
- Fractional Occupation Numbers: The pseudo-Fractional Occupation Number method allows orbital occupation numbers around the Fermi level to assume non-integer values following a Fermi-Dirac distribution: nₚ = (1 + e^(εₚ - εF)/kT)⁻¹ [81]. This includes multiple electron configurations in the same optimization, improving stability [81].
- Electron Smearing: Simulates a finite electron temperature using fractional occupation numbers to distribute electrons over near-degenerate levels [7]. The electronic temperature parameter should be kept as low as possible, potentially using multiple restarts with successively smaller smearing values [7].
- Level Shifting: Artificially raises the energy of unoccupied virtual orbitals to increase the HOMO-LUMO gap and reduce excessive mixing between occupied and virtual spaces [1] [83]. While effective for convergence, this technique can give incorrect values for properties involving virtual orbitals, such as excitation energies and NMR shifts [7].
A representative input example for Q-Chem demonstrates the application of fractional occupations for a platinum cluster system [81]:
Open-Shell Configurations
Open-shell systems, particularly transition metal complexes, present unique challenges for SCF convergence:
- Spin Unrestricted Formalism: Essential for proper description of open-shell configurations, though spin-restricted open-shell Hartree-Fock (ROHF) is also available for high-spin systems [82].
- Spin Multiplicity Verification: Ensuring the correct spin multiplicity is specified is crucial, as improper spin settings represent a common source of convergence failure [7].
- Convergence from Ionic States: For problematic neutral open-shell systems, first converging the calculation for a closed-shell ion (cation or anion) and then using the resulting orbitals as an initial guess for the neutral system can be effective [83].
- Configuration-Averaged HF (CAHF): Available in Molpro, this method yields orbitals equivalent to state-averaged CASSCF calculations averaging over all states of possible spin manifolds in a chosen active space [62].
Transition Metal Complexes
Systems containing transition metals combine challenges from both small-gap and open-shell scenarios:
- Enhanced Convergence Criteria: Tightening SCF convergence tolerances (e.g.,
TightSCFin ORCA) with TolE of 1e-8 and TolRMSP of 5e-9 provides greater resolution in the convergence process [9]. - Conservative DIIS Parameters: Using more DIIS expansion vectors (e.g., N=25) with delayed DIIS onset (Cyc=30) and reduced mixing parameters (Mixing=0.015) can provide more stable, albeit slower, convergence [7].
- Damping and Shift Techniques: Applying damping factors (e.g., 0.5) in initial cycles combined with level shifts (200-500 cm⁻¹) can stabilize early iteration behavior [1] [83].
Implementation Protocols Across Computational Packages
ADF Implementation
The ADF package offers multiple SCF convergence accelerators and detailed parameter control [7]:
This configuration represents a "slow but steady" approach for difficult systems, with increased DIIS vectors, delayed DIIS onset, and reduced mixing parameters [7].
Q-Chem Fractional Occupation Protocol
Q-Chem's pseudo-Fractional Occupation Number method implementation includes several control parameters [81]:
- FONTSTART/FONTEND: Electronic temperature in Kelvin (default 1000 K)
- FON_NORB: Number of orbitals above and below Fermi level for fractional occupancy (default 4)
- FONETHRESH: DIIS error threshold for freezing occupations (default 4, corresponding to 10⁻⁴)
PySCF Convergence Control
PySCF provides extensive SCF customization through Python scripting [1]:
ORCA Convergence Tolerances
ORCA implements predefined convergence levels with associated tolerance suites [9]:
Table 3: ORCA SCF Convergence Tolerance Settings
| Criterion | LooseSCF | NormalSCF | TightSCF | VeryTightSCF |
|---|---|---|---|---|
| TolE (Energy) | 1e-5 | 1e-6 | 1e-8 | 1e-9 |
| TolRMSP (Density RMS) | 1e-4 | 1e-6 | 5e-9 | 1e-9 |
| TolMAXP (Max Density) | 1e-3 | 1e-5 | 1e-7 | 1e-8 |
| TolErr (DIIS Error) | 5e-4 | 1e-5 | 5e-7 | 1e-8 |
| Best For | Preliminary scans | Standard applications | Transition metals | High-precision properties |
Gaussian Practical Troubleshooting
For Gaussian users experiencing SCF convergence issues, practical solutions include [83]:
- SCF=NoVarAcc: Disables automatic grid reduction, helpful for calculations with diffuse functions
- SCF=NoIncFock: Turns off incremental Fock matrix construction, preventing approximation-induced convergence issues
- SCF=QC: Activates quadratic convergence algorithm at higher computational cost
- SCF=VShift=300-500: Applies level shifting of 300-500 cm⁻¹ to increase HOMO-LUMO gap
- Int=UltraFine: Uses finer integration grid, particularly important for Minnesota functionals
Visualization of SCF Convergence Workflows
The following workflow diagrams illustrate structured approaches to SCF convergence problems for challenging systems.
The Scientist's Toolkit: Essential Computational Reagents
Table 4: Key Computational Parameters and Their Functions
| Parameter/Technique | Function | Typical Values | Applicable Systems |
|---|---|---|---|
| Mixing Parameter | Controls Fock matrix update aggressiveness | 0.01-0.20 (default ~0.2) | All systems, particularly oscillating cases [7] |
| Level Shift | Increases HOMO-LUMO gap artificially | 0.1-0.5 Hartree | Small-gap systems, metals [1] [83] |
| Damping Factor | Stabilizes initial iterations | 0.3-0.8 | Diverging systems [1] |
| DIIS Vectors (N) | Number of previous Fock matrices for extrapolation | 8-25 (default ~10) | All systems, particularly unstable cases [7] |
| Electronic Temperature | Controls fractional occupation smearing | 300-5000 K | Metallic systems, small-gap semiconductors [81] |
| Integration Grid | Numerical integration accuracy | Fine/UltraFine (Gaussian) | DFT with difficult functionals [83] |
| Convergence Tolerance | Controls when SCF is considered converged | Loose to Extreme | Application-dependent [9] |
Successfully converging SCF calculations for challenging systems requires a systematic approach that combines physical insight with mathematical stabilization techniques. The most effective strategies begin with ensuring physical realism in the molecular geometry and appropriate spin specification before applying more advanced computational techniques [7]. For systems with small HOMO-LUMO gaps, fractional occupation methods and electron smearing provide the most physically motivated solution [81]. For oscillating systems, stabilization through damping and conservative DIIS parameters often proves effective [7] [1]. Most importantly, persistence and systematic testing of multiple approaches is essential, as difficult systems frequently require combining several techniques to achieve convergence. The protocols and methodologies outlined in this guide provide a comprehensive foundation for addressing even the most challenging SCF convergence scenarios in advanced electronic structure research.
The Self-Consistent Field (SCF) iterative process is a cornerstone computational method in electronic structure theory, forming the basis for both Hartree-Fock (HF) and Kohn-Sham Density Functional Theory (DFT) calculations. In this procedure, the ground-state wavefunction is expressed as a single Slater determinant of molecular orbitals, and the total electronic energy is minimized subject to orbital orthogonality constraints. This minimization leads to the SCF equation F C = S C E, where F is the Fock matrix, C contains the molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital eigenenergies [1]. The solution to this equation must be found self-consistently since the Fock matrix itself depends on the occupied orbitals through the density matrix.
Despite its formal elegance, the SCF process often faces significant convergence challenges in practical implementations, particularly for systems with metallic character, small HOMO-LUMO gaps, or complex electronic structures. These challenges manifest as oscillatory behavior during iterations, convergence to saddle points rather than true minima, or complete failure to converge. To address these issues, computational scientists have developed three powerful classes of techniques: smearing methods, Fermi broadening, and second-order convergence algorithms. These approaches have become essential tools for researchers investigating metallic systems, transition metal complexes, and other challenging molecular structures in materials science and drug development research.
Smearing Methods in SCF Calculations
Theoretical Foundation and Physical Rationale
Smearing methods address a fundamental challenge in SCF calculations for metallic systems: the discontinuity in occupation numbers at the Fermi surface where occupancies abruptly jump from 1 to 0 [84]. In periodic systems, the electron density is computed through integration over the Brillouin zone, which in practice requires summation over a finite set of k-points [84]. For metals, where bands cross the Fermi level and are only partially occupied, this discontinuity necessitates a prohibitively large number of k-points to achieve converged results.
The core concept of smearing involves replacing the discontinuous integer occupation numbers with a smooth function that varies gradually from 1 to 0 near the Fermi level [84]. From a physical perspective, smearing can be understood as occupying states according to a smooth distribution function, which helps avoid numerical problems arising from finite sampling of the Brillouin zone and specific properties of the investigated system [85]. In metallic systems with relatively flat bands at the Fermi energy, the absence of smearing can cause bands to be completely occupied in one iteration and completely unoccupied in the next as bands shift during the self-consistency cycle [85]. This leads to strong oscillations in the charge density between iterations, severely impeding convergence to a self-consistent solution.
Mathematical Formalism of Smearing
Mathematically, smearing is implemented by replacing the delta function in the density of states expression with a smeared function of finite width [84]. The generalized occupation function is given by:
[f(\epsilon) = \int_{-\infty}^{\mu} \tilde{\delta}(x - \epsilon) dx]
where μ is the Fermi level and (\tilde{\delta}) is the smearing function with width determined by a broadening parameter σ [84]. This formulation leads to the minimization of a generalized free energy functional rather than the internal energy functional, with the generalized temperature determined by the broadening parameter σ [84].
Table 1: Comparison of Key Smearing Methods in SCF Calculations
| Method | Mathematical Form | Key Properties | Optimal Use Cases |
|---|---|---|---|
| Fermi-Dirac | ( f{i\mathbf{k}} = \frac{1}{e^{(\epsilon{i\mathbf{k}} - \mu)/\sigma} + 1} ) | Physical meaning (electronic temperature), positive occupations | Finite-temperature calculations; conservative approach |
| Gaussian | Gaussian smearing function | No negative occupations, non-physical broadening | General metallic systems requiring stability |
| Methfessel-Paxton | Finite-order polynomial expansion | Possible negative occupations, fast convergence | Ground-state metallic systems (forces/stress) |
| Cold Smearing | Specially designed asymmetric function | No negative occupations, minimal free energy dependence | Accurate forces and structural properties |
Practical Implementation and System-Dependent Selection
The implementation of smearing methods requires careful selection of both the smearing function and the broadening parameter. For systems with a band gap, such as semiconductors, insulators, and molecules, either Fermi-Dirac or Gaussian smearing with a low broadening (approximately 0.01 eV) is recommended [84]. For metallic systems, Methfessel-Paxton or cold smearing with the largest possible broadening that maintains small entropy contributions to the free energy typically yields optimal results [84].
The significant computational advantage of smearing is demonstrated in the case of bulk aluminum, where using σ = 0.43 eV reduced the number of k-points needed for 1 meV convergence from 15,625 to 2,197—a seven-fold acceleration [84]. For ground-state calculations, the extrapolation formula ( E_{\sigma \to 0}(\sigma) = \frac{1}{2} [E(\sigma) + F(\sigma)] ) enables accurate determination of the zero-temperature energy even when using large broadenings [84].
Diagram 1: Decision workflow for selecting smearing methods in SCF calculations
Fermi Broadening in Electronic Structure Calculations
Fundamental Principles and Temperature Effects
Fermi broadening represents a specific implementation of smearing that employs the Fermi-Dirac distribution to determine orbital occupations. This approach has a clear physical interpretation: it corresponds to treating the electronic system at a finite temperature T, where σ = k₋B⁻T defines the broadening width [84]. The Fermi-Dirac distribution naturally introduces smooth occupation transitions around the Fermi level, with the derivative of the distribution function having a full width at half maximum of approximately 3.5k₋B⁻T [86].
In the context of Fermi liquid theory, which describes the normal state of conduction electrons in most metals at low temperatures, the finite lifetime of quasiparticles near the Fermi surface provides a physical justification for broadening [87]. The condition ħ/τ << k₋B⁻T for quasiparticles ensures that their energy remains well-defined despite scattering processes, supporting the use of temperature-based broadening in calculations [87].
Implementation in Quantum Chemistry Packages
Major quantum chemistry packages implement Fermi broadening as a convergence accelerator. In Gaussian, the SCF=Fermi keyword requests "temperature broadening during early iterations, combined with CDIIS and damping" [15]. This approach specifically helps with difficult convergence cases by preventing oscillatory behavior in the initial SCF cycles. The implementation typically combines Fermi broadening with damping and level shifting, providing multiple stabilization mechanisms during the critical early iterations [15].
PySCF provides smearing capabilities through fractional occupancy settings, allowing users to employ Fermi-Dirac smearing for challenging systems [1]. The package offers examples demonstrating how smearing can be activated to assist SCF convergence, particularly for systems with small HOMO-LUMO gaps or metallic character [1].
Practical Guidelines and Parameter Selection
When implementing Fermi broadening, the broadening parameter should be selected based on system characteristics. For most systems, a value between 0.1-0.3 eV provides sufficient smoothing without introducing excessive temperature effects. For ground-state property calculations, it is essential to extrapolate to zero broadening using established techniques [84].
The electronic entropy term in the free energy functional becomes significant when using Fermi broadening, with S representing the electronic entropy in the expression F[n] = E[n] - TS [84]. This entropy contribution must be considered when evaluating the validity of results, particularly for low-temperature systems.
Table 2: Fermi Broadening Parameters for Different System Types
| System Type | Recommended σ (eV) | Physical Temperature Equivalent (K) | Key Considerations |
|---|---|---|---|
| Metals | 0.1-0.5 | 1160-5800 | Use larger values for k-point convergence; extrapolate to σ=0 |
| Semiconductors/Insulators | 0.01-0.05 | 116-580 | Minimal broadening to avoid artificial metallization |
| Transition Metal Complexes | 0.05-0.2 | 580-2320 | Address challenging convergence with intermediate values |
| Finite-Temperature Studies | 0.026-0.26 | 300-3000 | Match broadening to actual simulation temperature |
Second-Order Convergence Methods
Theoretical Basis of Second-Order Optimization
Second-order SCF methods leverage mathematical optimization principles to achieve superior convergence characteristics compared to conventional first-order approaches. While standard SCF algorithms typically employ Direct Inversion in the Iterative Subspace (DIIS) for convergence acceleration [1], second-order methods utilize approximate or exact second derivatives of the energy with respect to orbital rotations to achieve quadratic convergence [88].
The fundamental advantage of second-order methods lies in their convergence rate. Near the solution, the error decreases quadratically rather than linearly, dramatically reducing the number of iterations required to reach convergence. The mathematical foundation involves solving the Newton-Raphson equations for orbital optimization, which require computation of the orbital Hessian—the second derivative of the energy with respect to orbital rotations [88].
Implementation Variants and Computational Approaches
Several implementations of second-order methods have been developed, each with distinct computational characteristics:
Quadratically Convergent (QC) SCF: This approach, available in Gaussian through the SCF=QC keyword, employs Newton-Raphson steps when close to convergence and linear searches when farther away [15]. The method is more reliable than conventional DIIS but computationally more expensive, making it particularly valuable for difficult cases where standard approaches fail.
Second-Order SCF (SOSCF): PySCF implements SOSCF through the co-iterative augmented Hessian (CIAH) method [1]. This can be invoked by decorating SCF objects with the newton() method. The implementation achieves quadratic convergence in orbital optimization while managing computational costs through iterative techniques.
Approximate Second-Order Methods: As described by Chaban, Schmidt, and Gordon, approximate second-order methods use diagonal guesses for the orbital Hessian with iterative updates [88]. This approach reduces computational costs while maintaining improved convergence characteristics compared to first-order methods, particularly for multiconfigurational SCF wavefunctions.
Comparative Performance and Practical Applications
For closed and open shell SCF calculations, approximate second-order methods can rival conventional DIIS in convergence speed [88]. For multiconfigurational SCF wavefunctions, these methods may require more iterations than exact second-order approaches, but each iteration is substantially faster, leading to more efficient overall performance [88].
The SCF=YQC algorithm in Gaussian represents a hybrid approach that begins with steepest descent, transitions to scaled steepest descent, then switches to regular SCF, using the quadratic algorithm only if the regular SCF fails to converge [15]. This adaptive strategy balances efficiency and reliability for very large molecules.
Diagram 2: Hierarchy of SCF convergence acceleration strategies
Integrated Approaches and Advanced Protocols
Combined Methodologies for Challenging Systems
For particularly challenging systems, integrated approaches combining multiple convergence acceleration techniques often yield the best results. A typical protocol might begin with Fermi broadening and damping during early SCF cycles to establish stable convergence patterns, then transition to more aggressive second-order methods once the electronic structure has partially stabilized.
The Gaussian package explicitly combines Fermi broadening with CDIIS and damping when the SCF=Fermi keyword is specified [15]. This integrated approach addresses convergence challenges through multiple simultaneous mechanisms: Fermi broadening smooths occupation transitions, damping prevents large oscillatory updates, and CDIIS provides optimal extrapolation based on previous iterations.
Specialized Protocols for Complex Electronic Structures
Metallic Systems Protocol:
- Initialize with superposition of atomic potentials or atomic density matrix [1]
- Employ Methfessel-Paxton or cold smearing with σ = 0.1-0.5 eV [84]
- Use a moderate k-point grid (e.g., 13×13×13 for simple metals) [84]
- Apply DIIS extrapolation with damping for initial 5-10 cycles [1]
- For final convergence, disable smearing and extrapolate to σ=0 if accurate ground-state energies are required [84]
Open-Shell and Multiconfigurational Systems Protocol:
- Utilize approximate second-order methods for orbital optimization [88]
- Implement level shifting to increase occupied-virtual orbital gaps [1]
- Consider fractional occupancy or smearing if near-degeneracies persist [1]
- Perform stability analysis to ensure true ground state convergence [1]
Nanoscale and Surface Systems Protocol:
- Employ density matrix initialization from smaller calculations or fragments [1]
- Use Fermi-Dirac smearing with σ ≈ 0.01-0.05 eV to handle small band gaps [84]
- Implement SOSCF for final convergence [1]
- Verify results with multiple initial guesses to detect meta-stable states [1]
Table 3: Research Reagent Solutions for SCF Convergence Challenges
| Tool/Resource | Function/Purpose | Implementation Examples |
|---|---|---|
| Smearing Functions | Smooth orbital occupations near Fermi level | Fermi-Dirac, Gaussian, Methfessel-Paxton, Cold [84] |
| DIIS Algorithms | Extrapolate Fock matrix from previous iterations | EDIIS, CDIIS, ADIIS [1] |
| Second-Order Solvers | Achieve quadratic convergence near solution | Newton-Raphson, CIAH, approximate Hessian [88] |
| Damping/Level Shift | Stabilize early SCF iterations | Dynamic damping, level shifting (0.001-0.1 Hartree) [1] |
| Initial Guess Methods | Provide starting point for SCF iterations | MINAO, Hückel, superposition of atomic potentials [1] |
| Stability Analysis | Verify solution is true minimum, not saddle point | Internal/external stability checks [1] |
Smearing methods, Fermi broadening, and second-order convergence algorithms represent essential components of the modern computational chemist's toolkit for addressing SCF convergence challenges. These techniques enable the successful application of electronic structure theory to increasingly complex systems in materials science, catalysis, and pharmaceutical development.
Smearing methods, particularly Fermi-Dirac and specialized approaches like Methfessel-Paxton and cold smearing, provide robust solutions for metallic systems and cases with near-degeneracies. Fermi broadening offers a physically motivated approach to convergence acceleration, with well-defined temperature dependencies. Second-order methods guarantee convergence for challenging cases where conventional approaches fail, though at increased computational cost per iteration.
The optimal application of these techniques requires understanding their theoretical foundations, practical implementations, and appropriate domain-specific protocols. As computational methods continue to evolve toward more complex and physically accurate simulations, these advanced SCF convergence strategies will remain indispensable for researchers pushing the boundaries of electronic structure calculation capabilities. Future developments will likely focus on improved adaptive algorithms that automatically select optimal convergence strategies based on system characteristics, further enhancing the reliability and applicability of SCF methods across diverse scientific domains.
Ensuring SCF Solution Quality: Validation, Analysis, and Method Selection
The self-consistent field (SCF) iterative process is a fundamental computational method for solving Hartree-Fock (HF) and Kohn-Sham density functional theory (KS-DFT) equations in electronic structure calculations. In both HF and KS-DFT, the ground-state wavefunction is expressed as a single Slater determinant, and the total electronic energy is minimized subject to orbital orthogonality constraints [1]. This minimization leads to the SCF equation F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, S is the atomic orbital overlap matrix, and E is a diagonal matrix of orbital eigenenergies [1].
A significant challenge in SCF calculations is that the iterative process may converge to solutions that do not represent the true electronic ground state. Even when the SCF converges such that the orbital gradient vanishes and the equation F C = S C E is satisfied, the obtained wavefunction may correspond to a saddle point on the electronic energy landscape rather than a local minimum [1]. In such cases, the energy can be decreased by perturbing the orbitals away from the saddle point, indicating that the wavefunction is unstable.
Stability analysis is essential because SCF solutions can converge to:
- True local minima: Stable solutions where all eigenvalues of the electronic Hessian are positive
- Saddle points: Unstable solutions where at least one eigenvalue of the electronic Hessian is negative [89]
- Excited states: Higher-energy stationary points that may be physically meaningful for calculating excited-state properties [90] [91]
The detection and correction of these saddle points is crucial for obtaining reliable ground-state energies and properties, and also provides a foundation for direct calculation of excited electronic states using methods such as ΔSCF [90] [92].
Theoretical Foundation of SCF Stability
Mathematical Formulation of SCF Stability
The stability of an SCF solution is determined by the electronic Hessian matrix, which contains the second derivatives of the energy with respect to orbital rotations. For both HF and DFT methods, the SCF stability analysis evaluates the electronic Hessian at the converged solution to determine its eigenvalues [89]. The sign of these eigenvalues determines the nature of the stationary point:
- If all eigenvalues are positive, the solution represents a local minimum on the electronic energy surface
- If one or more eigenvalues are negative, the solution corresponds to a saddle point [89]
Instabilities are conventionally classified as either:
- Internal instabilities: The SCF has converged to an excited state instead of the ground state
- External instabilities: The energy can be decreased by loosening constraints on the wavefunction, such as allowing restricted Hartree-Fock (RHF) orbitals to transform into unrestricted Hartree-Fock (UHF) orbitals [1]
Physical Interpretation of Saddle Points
Saddle points in SCF calculations typically arise in systems with strong electron correlation, near-degeneracies, or symmetry constraints that prevent the system from reaching the true ground state. Common physical scenarios include:
- Stretched bonds in diatomic molecules where symmetry constraints lead to restricted solutions instead of the preferred unrestricted ones [89]
- Systems with small HOMO-LUMO gaps where orbital energy degeneracy or near-degeneracy creates multiple stationary points [1]
- Open-shell systems where spin symmetry constraints lead to unstable solutions [1]
- Charge-transfer systems where delocalization error in DFT functionals promotes unphysical solutions [92]
From a theoretical perspective, recent work by Yang and Ayers has established that stationary solutions of the same energy functional whose minimum corresponds to the ground state can yield excited-state energies and electron densities, providing the theoretical foundation for ΔSCF calculations of excited states [90].
Detecting SCF Instabilities: Methodologies and Protocols
Stability Analysis Algorithms
Stability analysis algorithms compute the lowest eigenvalues of the electronic Hessian to determine if negative eigenvalues exist. The implementation in ORCA, for example, limits analysis to RHF/RKS in the space of UHF/UKS or UHF/UKS in the space of UHF/UKS [89].
The electronic Hessian determination is structurally comparable to the TDHF/CIS/TDDFT procedure, employing similar iterative algorithms to find the lowest eigenpairs [89]. A typical stability analysis calculation involves:
- Converging an SCF solution using standard iterative procedures
- Constructing the electronic Hessian or implementing an iterative algorithm to find its lowest eigenvalues
- Analyzing the eigenvalue spectrum to identify negative eigenvalues
- Classifying the instability (internal vs. external) based on the nature of the corresponding eigenvectors
Practical Implementation in Quantum Chemistry Codes
Different quantum chemistry packages implement stability analysis with varying features and capabilities:
Table 1: Stability Analysis Implementation in Quantum Chemistry Codes
| Program | Key Features | Available Stability Types | Key References |
|---|---|---|---|
| PySCF | Detects both internal and external instabilities | Internal and external instabilities for RHF/RKS and UHF/UKS | [1] |
| ORCA | Evaluates electronic Hessian; can automatically restart UHF if unstable | RHF/RKS in space of UHF/UKS; UHF/UKS in space of UHF/UKS | [89] |
| ADF | Stability analysis for various Hamiltonian types | Not specified in available sources | [93] |
The following workflow diagram illustrates the general procedure for performing stability analysis:
Step-by-Step Experimental Protocol
For researchers implementing stability analysis in ORCA, the following detailed protocol can be employed:
Perform initial SCF calculation with careful convergence:
Request stability analysis in the input block:
Analyze output for negative eigenvalues indicating instability
If unstable, employ the modified guess orbitals generated by the stability analysis to restart the SCF procedure
Verify the final solution is stable by repeating the stability analysis
Key parameters for the stability analysis in ORCA include:
STABNRoots: Number of eigenpairs sought (typically 3-5)STABMaxDim: Davidson expansion space = MaxDim × NRootsSTABDTolandSTABRTol: Convergence criteria (typically 0.0001)STABRestartUHFifUnstable: Automatically restart UHF if unstable [89]
Correcting Unstable SCF Solutions
Advanced SCF Convergence Algorithms
When unstable SCF solutions are detected, several advanced algorithms can be employed to achieve convergence to stable solutions:
DIIS (Direct Inversion in Iterative Subspace): The default method in many codes that extrapolates the Fock matrix using previous iterations by minimizing the norm of the commutator [F, PS] where P is the density matrix [1]
Second-order SCF (SOSCF): Achieves quadratic convergence in orbital optimization using the co-iterative augmented Hessian (CIAH) method [1]
Optimal Damping Algorithm (ODA): Ensures monotonic energy decrease through an automatic line search procedure [94] [78]
Adaptive damping algorithms: Recently developed methods that automatically adjust damping parameters in each SCF step based on a line search [94]
Specific Techniques for Challenging Cases
Table 2: Techniques for Correcting Unstable SCF Solutions
| Technique | Methodology | Applicable Systems | Key References |
|---|---|---|---|
| Level shifting | Increases gap between occupied and virtual orbitals | Systems with small HOMO-LUMO gaps | [1] |
| Fractional occupations | Smears occupations according to temperature function | Metallic systems, small-gap systems | [1] |
| Damping | Mixes old and new Fock matrices | Early SCF iterations, charge-sloshing instabilities | [1] [94] |
| Improved initial guess | Uses better starting orbitals/density | All systems, particularly transition metals | [1] [51] |
| Symmetry breaking | Allows orbitals to break spatial/spin symmetry | Stretched bonds, open-shell systems | [89] |
The following diagram illustrates the decision process for selecting appropriate correction techniques:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for SCF Stability Analysis
| Tool/Resource | Function/Purpose | Implementation Details |
|---|---|---|
| Stability analysis | Detects saddle points in SCF solutions | Implemented in PySCF, ORCA, ADF; computes electronic Hessian eigenvalues [1] [89] |
| DIIS variants | Accelerates SCF convergence | EDIIS, ADIIS; minimizes commutator [F, PS] [1] |
| Second-order SCF | Provides quadratic convergence | Co-iterative augmented Hessian method; invoked via .newton() in PySCF [1] |
| OpenOrbitalOptimizer | Reusable library for SCF calculations | Implements DIIS, EDIIS, ADIIS, ODA; written in C++ [78] |
| Adaptive damping | Automatically adjusts damping parameters | Based on line search; requires no user input [94] |
| Initial guess methods | Provides better starting points | minao, atom, huckel, vsap; critical for difficult systems [1] |
Connection to Excited-State Calculations
ΔSCF Methods for Excited States
The same saddle point solutions that pose problems for ground-state calculations can be intentionally targeted for calculating excited states. The ΔSCF (delta SCF) method involves converging the SCF procedure to stationary points that represent excited electronic states rather than the ground state [90] [92].
Recent theoretical work has established the foundation for ΔSCF calculations, showing that while the minimum of the energy functional corresponds to the ground state energy, the stationary solutions yield excited-state energies and electron densities [90]. This provides a rigorous theoretical justification for what was previously an empirically successful approach.
Several variants of ΔSCF have been developed to facilitate convergence to target excited states:
- Maximum Overlap Method (MOM): Maximizes overlap between occupied orbitals on successive SCF iterations to prevent variational collapse to the ground state [91] [92]
- Initial Maximum Overlap Method (IMOM): Enhanced version of MOM with improved convergence [92]
- σ-SCF method: Alternative approach for targeting specific excited states [92]
- State-Targeted Energy Projection (STEP): Projects out lower energy states to facilitate convergence to higher states [92]
- Restricted Open-Shell Kohn-Sham (ROKS): For low-spin excited states, based on Ziegler's sum method [92]
Benchmarking ΔSCF Performance
Recent comprehensive benchmarks of ΔSCF methods have evaluated their performance for calculating excited-state properties such as dipole moments. Key findings include:
- ΔSCF provides reasonable accuracy for certain doubly excited states, which are not accessible to conventional TDDFT calculations [92]
- For charge-transfer states, ΔSCF may suffer from DFT overdelocalization error more severely than TDDFT [92]
- In push-pull systems like donor-acceptor-substituted polyenes, this overdelocalization can lead to beneficial error cancellation [92]
- On average, ΔSCF data do not necessarily improve on TDDFT results but offer increased accuracy in certain pathological cases [92]
Protocol for Excited-State Calculations via ΔSCF
For researchers interested in applying ΔSCF for excited-state calculations, the following protocol is recommended:
- Select appropriate initial orbitals that approximate the target excited state
- Employ a ΔSCF variant (MOM, IMOM, etc.) to maintain the desired state during iterations
- Perform stability analysis on the converged solution to confirm it represents the desired stationary point
- Calculate excited-state properties using the same methodology as for ground states
- Validate results against experimental data or higher-level theoretical methods when possible
For excited-state geometry optimizations, programs like ADF implement specialized keywords:
The EIGENFOLLOW option attempts to follow the eigenvector during geometry optimization, which is particularly important when states cross [93].
Stability analysis is an essential component of robust SCF calculations, enabling researchers to distinguish true ground-state solutions from saddle points and excited states. The methodologies described in this guide—from basic stability analysis to advanced convergence algorithms and ΔSCF techniques—provide a comprehensive toolkit for addressing these challenges.
As quantum chemical applications expand to more complex systems including transition metal complexes, extended materials, and excited-state processes, the importance of stability analysis only grows. Recent developments in automatic adaptive algorithms [94] and reusable libraries like OpenOrbitalOptimizer [78] promise to make these powerful techniques more accessible to non-specialists, potentially transforming stability analysis from a specialist's tool to a routine component of electronic structure calculations.
The integration of stability analysis with excited-state methodologies like ΔSCF highlights the dual nature of saddle points—while they represent failures in ground-state calculations, they become valuable targets for excited-state studies. This unifying perspective underscores the fundamental continuity of electronic structure theory across ground and excited states.
The self-consistent field (SCF) method is a cornerstone computational technique in quantum chemistry, forming the basis for both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (DFT). In this iterative procedure, the electronic wavefunction is expressed as a single Slater determinant of molecular orbitals (MOs), and the total electronic energy is minimized subject to orbital orthogonality constraints [1]. This approach effectively describes electrons as independent particles interacting through a mean field, leading to the fundamental SCF equation F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, E is a diagonal matrix of orbital eigenenergies, and S is the atomic orbital overlap matrix [1].
Despite its widespread success, a significant challenge in SCF methodology arises from the potential convergence to unstable wavefunctions—solutions that represent saddle points rather than true energy minima in the space of possible wavefunctions [95]. At convergence, any SCF energy will be at a stationary point with respect to changes in the MO coefficients, but this stationary point is not guaranteed to be an energy minimum [95]. When the energy can be lowered by perturbing the wavefunction in some direction, the solution is deemed unstable.
Understanding and classifying these instabilities is crucial for researchers relying on quantum chemical computations, particularly in drug development where accurate predictions of molecular properties, reaction mechanisms, and spectroscopic parameters depend on obtaining the correct electronic ground state. This guide provides an in-depth examination of wavefunction instabilities, their diagnostic methods, and practical resolution protocols within the context of SCF iterative processes.
Theoretical Foundation of SCF Instabilities
Mathematical Formalism of Instabilities
Wavefunction instabilities originate from the mathematical structure of the SCF equations. The stability of an SCF solution is determined by the electronic Hessian (second derivative of the energy with respect to orbital rotation parameters). A stable solution requires all eigenvalues of this Hessian to be positive [89]. If one or more negative eigenvalues exist, the solution represents a saddle point and is unstable [89].
The most general form for spin orbitals is:
χᵢ(𝐫,ζ) = ψᵢα(𝐫)α(ζ) + ψᵢβ(𝐫)β(ζ)
where ψᵢα and ψᵢβ are complex-valued functions of the Cartesian coordinates 𝐫, and α and β are spin eigenfunctions [95]. Practical SCF calculations typically apply several constraints to this general form:
- Spin restriction: Assuming spin orbitals depend only on one spin function (α or β)
- Reality constraint: Using real rather than complex functions
- Spatial restriction: Requiring ψᵢα = ψᵢβ (restricted formalism) [95]
Each constraint potentially artificially stabilizes the wavefunction, and instability arises when relaxing these constraints leads to a lower energy solution.
Classification of Instabilities
SCF instabilities are systematically classified based on the constraints relaxed when lower-energy solutions are found:
Table 1: Classification of Wavefunction Instabilities
| Instability Type | Constraints Relaxed | Characteristic | Common Occurrence |
|---|---|---|---|
| Internal | None (within same symmetry) | Converged to excited state | Systems with small HOMO-LUMO gaps |
| External | Symmetry/spin constraints | Lower-energy solution exists with different symmetry | Singlet diradicals, bond dissociation |
| Restricted → Unrestricted | Spin restriction (RHF→UHF) | Spatial parts of α and β orbitals differ | Open-shell systems described with restricted orbitals |
| Real → Complex | Reality constraint | Complex orbitals yield lower energy | Systems with strong magnetic fields or spin-orbit coupling |
Internal instabilities occur when the SCF procedure converges to an excited state within the same symmetry constraints, rather than the true ground state [1]. This typically happens in systems with small HOMO-LUMO gaps where the orbital energy spectrum creates challenges for convergence algorithms.
External instabilities arise when the energy can be lowered by loosening constraints on the wavefunction, such as allowing restricted Hartree-Fock orbitals to transform into unrestricted orbitals [1] [95]. The restricted → unrestricted instability is particularly common in systems with significant spin polarization or during bond dissociation.
Diagnostic Methods for Wavefunction Instabilities
Stability Analysis Theory
Stability analysis involves computing the lowest eigenvalues of the electronic Hessian matrix with respect to orbital rotations [89]. If negative eigenvalues are found, the solution is unstable along the directions corresponding to the eigenvectors. Two primary approaches exist for evaluating the Hessian-vector products required in stability analysis:
- Analytical evaluation: Available for standard RHF/RKS and UHF/UKS cases with common functionals [95]
Finite-difference methods: Used when analytical Hessians are unavailable, employing the formula:
H b₁ = [∇E(X₀ + ξb₁) - ∇E(X₀ - ξb₁)] / 2ξ
where H is the Hessian matrix, b₁ is a trial vector, X₀ stands for the current stationary point, and ξ is the finite step size [95]
This finite-difference approach enables stability analysis for a wider range of theoretical methods, including those with non-local correlation (NLC) functionals where analytical second derivatives may be unavailable [95].
Practical Implementation in Quantum Chemistry Codes
Most major quantum chemistry packages implement stability analysis tools:
Q-Chem provides comprehensive stability analysis within its GENSCFMAN module, with options for multiple automated correction cycles (INTERNALSTABILITY_ITER) [95]. The implementation can handle various wavefunction types and automatically displaces MOs along the lowest-energy eigenvector with line search if instability is detected, followed by new SCF calculations until a stable solution is reached [95].
ORCA offers stability analysis via the STABPerform keyword, with options to control the number of roots sought (STABNRoots), convergence criteria (STABDTol, STABRTol), and orbital window for the analysis [89]. ORCA limits its analysis primarily to RHF/RKS in the space of UHF/UKS or UHF/UKS in the space of UHF/UKS [89].
PySCF allows detection of both internal and external instabilities through its stability analysis tools, with examples provided in the documentation [1]. The code offers flexibility in analyzing various wavefunction types and provides mechanisms for generating improved initial guesses from unstable solutions.
The following diagram illustrates the comprehensive diagnostic workflow for identifying and addressing wavefunction instabilities:
Diagram 1: Comprehensive workflow for diagnosing and addressing wavefunction instabilities in SCF calculations.
Quantitative Diagnostics and Thresholds
Beyond formal stability analysis, several quantitative metrics can help identify problematic wavefunctions:
- T1 and D1 diagnostics: Based on amplitudes of singles in the CCSD wavefunction, these metrics help assess multireference character that may lead to SCF instabilities [96]
- Orbital energy gaps: Small HOMO-LUMO gaps (<0.05 a.u.) often correlate with instability problems
- Density matrix oscillations: Significant oscillations in SCF iterations may indicate underlying instabilities
While these diagnostics provide valuable indicators, it's important to note that their relationship to predictable error bars is not always straightforward [96]. There are documented cases where coupled-cluster methods like CCSD(T) provide accurate results despite large values of these diagnostics [96].
Implications of Unstable Wavefunctions
Impact on Chemical Predictions
Unstable SCF solutions can significantly compromise the reliability of computational chemistry predictions in drug development applications:
Spin-state energetics: Incorrect relative energies between different spin states can lead to wrong assignments of ground states in transition metal complexes, which are common in metalloenzymes and catalysts [96]. Studies have shown that methods like CASPT2 with default parameters may overstabilize high-spin states by several kcal/mol compared to more accurate CCSD(T) benchmarks [96].
Reaction barrier heights: Unstable reference wavefunctions can introduce errors in calculated activation energies, affecting the prediction of reaction rates in metabolic pathways or drug metabolism studies.
Molecular properties: Dipole moments, polarizabilities, and spectroscopic constants computed from unstable wavefunctions may contain significant errors, reducing their utility in interpreting experimental data.
Non-covalent interactions: Weak intermolecular forces crucial to drug-receptor binding are particularly sensitive to the quality of the reference wavefunction.
Relationship to Electron Correlation
Wavefunction instabilities are intimately connected to electron correlation effects. The single-determinant approximation inherent in standard SCF methods fails to adequately capture:
- Static (non-dynamic) correlation: Important in systems with near-degeneracies, such as bond dissociation or diradicals [96]
- Dynamic correlation: The instantaneous correlation between electrons due to their Coulomb repulsion [96]
Multireference methods like CASSCF specifically address static correlation by using a combination of determinants but require careful selection of the active space [96]. The standard active space for transition metal complexes typically includes metal d orbitals along with correlating d' orbitals and ligand orbitals involved in covalent bonding with the metal [96].
Table 2: Computational Methods for Addressing Correlation-Related Instabilities
| Method Class | Representative Methods | Strengths | Limitations | Applicability |
|---|---|---|---|---|
| Single-Reference Correlation | CCSD(T), CCSD(T)-F12 | High accuracy for dynamic correlation, "gold standard" for many systems [96] | Fails for genuine multireference cases [96] | TM complexes near equilibrium |
| Multireference Wavefunction | CASSCF, RASSCF, DMRG | Handles static correlation explicitly [96] | Active space selection challenging [96] | Diradicals, bond dissociation |
| Multireference Perturbation Theory | CASPT2, NEVPT2 | Adds dynamic correlation to CASSCF [96] | Parameter sensitivity (e.g., IPEA shift) [96] | Spectroscopy, spin states |
| Hybrid Approaches | CASPT2/CC | Combines CASPT2 for valence correlation with CCSD(T) for outer-core correlation [96] | Computational complexity | High-accuracy TM complexes |
Resolution Protocols for Unstable Wavefunctions
Technical Approaches for SCF Convergence
When facing SCF convergence problems or identified instabilities, several technical strategies can be employed:
Advanced mixing algorithms: Beyond simple DIIS, methods like EDIIS [1], ADIIS [1], or Pulay mixing [79] can improve convergence. The damping factor (
SCF.Mixer.Weightin SIESTA) controls the percentage of the new density or Hamiltonian matrix used in each iteration [79].Level shifting: Artificially increasing the gap between occupied and virtual orbitals can stabilize the SCF procedure [1]. This approach is particularly useful for systems with small HOMO-LUMO gaps.
Fractional occupations: Assigning fractional occupancies to orbitals near the Fermi level can help achieve convergence in metallic systems or those with small band gaps [1].
Second-order SCF (SOSCF): Methods like the co-iterative augmented hessian (CIAH) approach can achieve quadratic convergence in orbital optimization [1].
Systematic Correction of Unstable Solutions
When stability analysis identifies an unstable wavefunction, systematic correction protocols should be employed:
Displacement along unstable modes: Modern implementations automatically displace orbitals along the direction of the lowest-energy eigenvector with line search [95]
Progressive constraint relaxation: Begin with the most constrained wavefunction (RHF) and progressively relax constraints (UHF, complex orbitals) until a stable solution is found
Alternative initial guesses: Poor initial guesses can lead to convergence on saddle points. Options include:
Multireference methods: For genuinely multireference systems, transition to CASSCF or DFT/multireference hybrid approaches
The following toolkit table summarizes essential resources for diagnosing and addressing wavefunction instabilities:
Table 3: Research Reagent Solutions for Wavefunction Instability Challenges
| Tool/Resource | Function | Implementation Examples | Key Parameters |
|---|---|---|---|
| Stability Analysis | Identifies internal/external instabilities | Q-Chem INTERNAL_STABILITY [95], ORCA STABPerform [89], PySCF stability check [1] | Number of roots, convergence tolerance |
| Advanced SCF Mixing | Accelerates and stabilizes convergence | SIESTA Pulay/Broyden [79], PySCF DIIS/EDIIS/ADIIS [1] | Mixing weight, history length |
| Orbital Modification | Escapes saddle point solutions | ORCA STABlambda [89], Q-Chem automated correction [95] | Displacement parameter, orbital window |
| Initial Guess Library | Provides alternative starting points | PySCF minao/atom/huckel/vsap [1] | Atomic density superposition |
| Multireference Methods | Handles strong correlation | CASSCF/CASPT2/NEVPT2 [96] | Active space selection |
Composite Protocols for Challenging Systems
For particularly challenging systems such as transition metal complexes or extended conjugated molecules, composite computational protocols have been developed:
Basis set incompleteness correction: Using explicitly correlated (F12) methods like CCSD(T)-F12 to minimize basis set errors with triple-ζ quality basis sets [96]
Core-valence separation: Splitting correlation effects into valence and outer-core contributions, as in the CASPT2/CC method [96]
Cost-effective protocols: Efficient partitioning of computational resources based on different energy contributions (nonrelativistic frozen-core CCSD, relativistic corrections, and triples corrections) with composite basis sets [96]
These protocols aim to achieve chemical accuracy (1-3 kcal/mol) in relative energies such as spin-state splinnings, which is crucial for predictive computational chemistry in drug development [96].
Wavefunction instability represents a fundamental challenge in SCF methodology with significant implications for the reliability of computational predictions in drug development research. Proper identification and treatment of both internal and external instabilities through systematic stability analysis and correction protocols are essential components of robust computational workflows. The continuing development of automated stability analysis tools and composite methods that efficiently balance computational cost with accuracy promises to enhance the reliability of quantum chemical calculations for complex pharmaceutical systems. As computational chemistry plays an increasingly important role in rational drug design, mastery of wavefunction validation techniques remains crucial for producing trustworthy theoretical predictions that can effectively guide experimental research.
The self-consistent field (SCF) method is a cornerstone of computational quantum chemistry, providing the fundamental framework for solving the electronic Schrödinger equation in density-functional theory (DFT) and related methods [97]. The SCF procedure involves an iterative optimization of the electronic structure of a molecule until the energy and electron density converge to a stable solution [69]. This process is formally a chaos-theory problem and is not guaranteed to converge; while algorithms like DIIS and ADIIS almost always converge in 10–30 steps for simple organic molecules, open-shell systems or metal complexes often prove trickier [98].
Benchmarking SCF results has become increasingly important as computational chemistry expands into complex molecular systems and high-throughput screening environments. Traditional SCF methods, while generally reliable, face challenges in convergence for difficult systems and can be computationally expensive for large molecules [69] [97]. This technical guide examines the integration of semi-empirical and machine learning approaches as complementary methods for accelerating SCF convergence, improving initial guesses, and providing reference data for benchmarking within the context of SCF procedure research.
Theoretical Foundation of SCF Methodology
The SCF Iterative Process
The fundamental goal of SCF methods is to solve the electronic Schrödinger equation for a molecular system through an iterative process [97]. In the independent-electron (mean-field) approximation, the wave function is represented as an antisymmetrized product of one-electron functions (molecular orbitals), typically expressed as a Slater determinant [97]. The optimum set of MOs is obtained by variationally minimizing the energy in the SCF approximation, leading to equations of the form:
where the Fock operator f^(i) for the i-th electron includes an effective potential υ_eff(i) that depends on the spin orbitals of the other electrons [97].
The second key approximation introduces an atomic orbital (AO) basis set, with MOs expressed as linear combinations of AOs [97]. This transforms the problem into the Roothaan-Hall matrix equation:
where 𝐅 is the Fock matrix, 𝐂 contains the molecular orbital coefficients, 𝐒 is the AO overlap matrix, and 𝜺 is a diagonal matrix of orbital energies [97].
SCF Convergence Challenges and Acceleration Methods
SCF convergence is regulated through various technical parameters that set the maximum number of iterations, convergence criteria, and iterative update methods [69]. The default settings in modern computational packages handle most cases, but challenging systems require specialized approaches:
Convergence Criteria: The commutator of the Fock matrix and the density matrix (P-matrix) serves as the primary error metric. Convergence is typically achieved when the maximum element falls below the threshold SCFcnv (default: 1e-6) and the matrix norm below 10*SCFcnv [69].
Acceleration Methods: Several algorithms improve SCF convergence [69]:
- DIIS (Direct Inversion in Iterative Space): The original Pulay DIIS scheme accelerates convergence using information from previous iterations.
- ADIIS+SDIIS: A hybrid method by Hu and Wang that combines advantages of different DIIS approaches.
- LIST (LInear-expansion Shooting Technique): A family of methods developed in Y.A. Wang's group.
- MESA: A comprehensive method combining multiple acceleration techniques.
- SOSCF: Second-order SCF, a slower but more robust algorithm used when standard methods fail [98].
Table 1: Key SCF Acceleration Methods and Their Applications
| Method | Key Features | Typical Applications | Performance Considerations |
|---|---|---|---|
| DIIS | Fast convergence using previous iterations | Standard organic molecules | Default in many codes; may fail for difficult systems |
| ADIIS+SDIIS | Adaptive combination of methods | Default in ADF software | Balanced approach for most systems |
| LIST | Linear-expansion techniques | Problematic convergence cases | Sensitive to number of expansion vectors |
| MESA | Combines multiple methods | Complex systems with diverse characteristics | Can disable specific components as needed |
| SOSCF | Second-order convergence | Open-shell systems, metal complexes | More robust but computationally expensive |
Semi-Empirical Methods for SCF Initialization and Benchmarking
Semi-Empirical Approximations in Quantum Chemistry
Semi-empirical methods bridge the gap between classical molecular mechanics and full first-principles calculations, providing a computationally efficient alternative for initial geometry optimization and SCF starting points [99]. These methods employ simplified quantum mechanical equations parameterized against experimental data or higher-level theoretical results, dramatically reducing computational cost while maintaining quantum mechanical accuracy for certain applications.
The GFN2-xTB (Geometry, Frequency, Non-covalent, extended Tight Binding) method has emerged as a particularly valuable semi-empirical approach for generating initial molecular geometries [99]. This method includes an explicit electronic treatment of valence electrons and captures essential non-covalent interactions—including hydrogen bonding, dispersion, and hyperconjugation—that are often inadequately described by generic force-field methods.
Integrated Workflows Combining Semi-Empirical and SCF Methods
Modern computational workflows increasingly integrate semi-empirical and ab initio SCF methods in a hierarchical approach [99]. The CHAOS (Computed High-Accuracy Observables and Sigma Profiles) database generation exemplifies this strategy, implementing a standardized multi-step procedure:
Initial Conformer Generation: Using distance-geometry embedding with the ETKDG algorithm followed by energy pre-screening with a universal force field (UFF) [99].
Semi-Empirical Refinement: Conformer sampling and optimization with GFN2-xTB Hamiltonian through the CREST (Conformer-Rotamer Ensemble Sampling Tool) package [99].
High-Level SCF Calculation: Final optimization and property calculation using DFT with the ωB97X-D functional and def2-TZVP basis set [99].
This workflow efficiently produces representative, low-energy gas-phase structures while avoiding unnecessary computational overhead, demonstrating how semi-empirical methods can enhance SCF workflows.
Diagram 1: Integrated workflow combining semi-empirical and SCF methods for efficient quantum-chemical calculations
Benchmarking SCF Against Semi-Empirical Methods
When benchmarking SCF results against semi-empirical approximations, several key metrics should be considered:
- Geometric Parameters: Bond lengths, angles, and dihedral angles compared between methods.
- Energetic Properties: Relative energies of conformers, reaction energies, and barrier heights.
- Electronic Properties: Dipole moments, orbital energies, and charge distributions.
- Computational Efficiency: Calculation time and resource requirements.
Semi-empirical methods typically provide qualitatively correct geometries at a fraction of the computational cost of full SCF calculations, making them excellent for initial screening and conformational analysis. However, for quantitative accuracy—particularly for properties sensitive to electron correlation—high-level SCF methods with appropriate density functionals remain essential.
Machine Learning Approaches in SCF Workflows
ML-Augmented SCF Convergence
Machine learning approaches offer promising alternatives for addressing SCF convergence challenges, particularly for difficult systems where traditional methods struggle. ML algorithms can predict optimal initial guesses for molecular orbitals, identify problematic systems before calculation, and even suggest optimal SCF parameters based on molecular features.
The application of ML to SCF convergence problems is particularly valuable for high-throughput computational screening, where manual intervention for problematic systems is impractical. By training models on previously calculated systems, ML approaches can learn to recognize molecular patterns associated with convergence difficulties and apply appropriate remedies automatically.
σ-Profiles as Molecular Descriptors for ML
σ-Profiles have emerged as powerful molecular descriptors for machine learning models in quantum chemistry and thermodynamic property prediction [99]. Derived from quantum-chemical calculations within the COSMO (Conductor-like Screening Model) framework, σ-profiles represent the molecular surface charge-density distribution and encode essential information on polarity and hydrogen-bonding characteristics [99].
The CHAOS database provides 53,091 consistent σ-profiles along with additional quantum-chemical descriptors, creating a valuable resource for training ML models [99]. This large-scale, internally consistent database addresses previous limitations in σ-profile availability and quality, which had constrained ML model development.
Table 2: Key Molecular Descriptors in the CHAOS Database for ML Applications
| Descriptor Category | Specific Properties | Applications in SCF/ML |
|---|---|---|
| Structural | Optimized Cartesian coordinates, rotational constants, symmetry numbers | Geometry optimization, conformational analysis |
| Electronic | Dipole moment, HOMO-LUMO gap, SCF energy, polarizability | Electronic property prediction, SCF initialization |
| Vibrational | Harmonic frequencies, IR intensities, zero-point energy | Spectroscopy, thermodynamic properties |
| NMR | Shielding constants, susceptibility tensors | Spectral prediction, electronic environment analysis |
| Solvation | σ-profiles, cavity information, solvation energies | Solvent selection, thermodynamic modeling |
Hybrid SCF-ML Workflows for Property Prediction
Machine learning models incorporating σ-profiles and other quantum-chemical descriptors have demonstrated significant improvements in predicting molecular and thermodynamic properties [99]. These hybrid approaches leverage the physical basis of quantum-chemical descriptors while benefiting from the pattern recognition capabilities of ML algorithms.
The integration of graph-based representations with traditional molecular descriptors has proven particularly powerful. Approaches such as Dual Graph Convolutional Networks (DGCN) can capture both local molecular structure and global topological relationships in chemical space, enhancing prediction accuracy for complex properties [99].
Diagram 2: Hybrid SCF-ML workflow for property prediction
Experimental Protocols for Benchmarking Studies
Standardized Benchmarking Methodology
To ensure consistent and reproducible benchmarking of SCF methods against semi-empirical and ML approaches, we propose the following standardized protocol:
System Selection: Curate a diverse set of molecular systems representing different chemical domains, including small organic molecules, transition metal complexes, and systems with known SCF convergence difficulties.
Reference Calculations: Perform high-level SCF calculations with established functional/basis set combinations (e.g., ωB97X-D/def2-TZVP) to generate reference data [99].
Semi-Empirical Calculations: Execute corresponding calculations with semi-empirical methods (GFN2-xTB, AM1, PM6) using consistent molecular geometries.
ML Predictions: Apply machine learning models trained on databases like CHAOS to predict properties of interest.
Statistical Analysis: Quantify deviations using appropriate metrics (MAE, RMSE, MAX errors) and identify systematic trends.
Protocol for SCF Convergence Acceleration Benchmarking
When specifically benchmarking SCF convergence acceleration methods:
Initialization: Start from consistent initial guesses (e.g., superposition of atomic densities or semi-empirical orbitals).
Method Comparison: Apply different acceleration schemes (DIIS, ADIIS, LIST, SOSCF) to identical systems.
Convergence Monitoring: Track the SCF error (commutator of Fock and density matrices) versus iteration count [69].
Performance Metrics: Record the number of iterations to convergence, computational time per iteration, and success rate for challenging systems.
Parameter Sensitivity: Test the sensitivity of each method to key parameters such as the number of DIIS vectors or mixing parameters.
Protocol for ML-Enhanced SCF Workflows
For benchmarking the integration of ML approaches in SCF procedures:
Training Set Construction: Curate a diverse set of molecules with known SCF convergence behavior and properties.
Descriptor Selection: Compute relevant molecular descriptors (σ-profiles, geometric, electronic) for all training instances [99].
Model Training: Implement appropriate ML algorithms (GCN, RF, SVM) predicting SCF convergence behavior or initial guesses.
Validation: Test trained models on hold-out sets of molecules not included in training.
Performance Assessment: Compare ML-enhanced SCF procedures against standard approaches in terms of convergence rate and computational efficiency.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for SCF Benchmarking Studies
| Tool/Resource | Type | Primary Function | Application in SCF Research |
|---|---|---|---|
| ADF | Software Suite | Quantum Chemical Calculations | SCF implementation with various acceleration methods [69] |
| Q-Chem | Software Suite | Ab Initio Quantum Chemistry | SCF algorithms with reduced scaling (O(N)) [97] |
| Gaussian 16 | Software Suite | Electronic Structure Modeling | DFT calculations for σ-profiles and descriptors [99] |
| CREST/GFN2-xTB | Semi-Empirical Tool | Conformer Sampling & Optimization | Initial geometry generation for SCF workflows [99] |
| CHAOS Database | Data Resource | σ-Profiles & Molecular Descriptors | Training ML models, benchmarking [99] |
| RDKit | Cheminformatics | Molecular Manipulation | Initial structure generation & processing [99] |
| DIIS | Algorithm | SCF Convergence Acceleration | Standard Pulay DIIS method [69] |
| SOSCF | Algorithm | SCF Convergence | Second-order method for difficult cases [98] |
| MESA | Algorithm | SCF Convergence | Combined acceleration approach [69] |
| DGCN | ML Architecture | Graph-Based Learning | Capturing local/global molecular associations [99] |
Benchmarking SCF results against semi-empirical and machine learning approaches reveals a complex landscape of trade-offs between computational efficiency, accuracy, and applicability across chemical space. Traditional SCF methods with appropriate acceleration techniques remain essential for quantitative accuracy, particularly for properties sensitive to electron correlation and subtle electronic effects. Semi-empirical methods provide excellent starting points for SCF calculations and efficient screening tools for large molecular sets. Machine learning approaches, particularly those leveraging consistent quantum-chemical descriptors like σ-profiles from comprehensive databases such as CHAOS, offer promising pathways for accelerating SCF convergence and predicting molecular properties with reduced computational cost.
The future of SCF methodology lies in intelligent hybrid approaches that strategically combine these methodologies—using ML for initial guesses and problem classification, semi-empirical methods for geometry preprocessing, and high-level SCF for final accurate determination of electronic properties. As benchmark datasets grow and algorithms improve, the integration of these complementary approaches will continue to expand the scope and accuracy of computational quantum chemistry across fundamental research and drug development applications.
The self-consistent field (SCF) method represents a cornerstone in computational quantum chemistry, enabling the prediction of molecular electronic structure and properties through an iterative optimization process. Within both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (KS-DFT), the SCF procedure minimizes the total energy with respect to the molecular orbitals, leading to a set of equations that must be solved self-consistently [1] [20]. This technical guide focuses on the verification of three critical properties derived from SCF calculations: dipole moments, orbital energies, and population analyses. These properties provide essential insights into molecular charge distribution, reactivity, and bonding characteristics, with significant implications for drug design and materials science.
The accuracy of property prediction depends critically on the convergence and stability of the SCF process. The fundamental SCF equation takes the form of a generalized eigenvalue problem, F C = S C E, where F is the Fock matrix, C is the matrix of molecular orbital coefficients, S is the overlap matrix of the atomic orbital basis set, and E is a diagonal matrix of orbital energies [1] [20]. The solution of this equation yields the molecular orbitals and their energies, which form the foundation for all subsequent property calculations discussed in this guide.
Theoretical Foundation of SCF Property Calculations
The SCF Iterative Process
The SCF method operates through a cyclic process wherein an initial guess of the molecular orbitals is progressively refined until convergence is achieved. This process begins with the construction of a core Hamiltonian, followed by the formation of the Fock matrix, which depends on the electron density. The matrix equation F C = S C E is solved to obtain improved orbitals, and the cycle repeats until the density matrix or total energy changes negligibly between iterations [1] [20].
A key challenge in SCF calculations lies in achieving convergence to a physically meaningful solution. The initial orbital guess significantly influences both convergence behavior and the final solution obtained. PySCF implements several guess strategies including 'minao' (superposition of atomic densities), 'atom' (superposition of atomic Hartree-Fock densities), 'huckel' (parameter-free Hückel method), and '1e' (one-electron Hamiltonian) [1]. For difficult cases, convergence accelerators such as direct inversion in iterative subspace (DIIS), damping, and level shifting are employed to stabilize the process [1].
Electronic Density and Property Derivation
In the SCF framework, the electron density serves as the fundamental variable for property calculation. For a single-determinant wavefunction, the electron density is constructed from the occupied molecular orbitals as follows:
$$ \rho(\vec{r}) = \sum{i}^{N} |\phii(\vec{r})|^2 = \sum{\mu\nu} P{\mu\nu} \chi{\mu}(\vec{r}) \chi{\nu}(\vec{r}) $$
where P is the density matrix, and χ are the basis functions [20]. This electron density forms the basis for calculating molecular properties, including the dipole moment, which can be obtained as the sum of nuclear and electronic contributions:
$$ \vec{\mu} = \vec{\mu}{nuc} + \vec{\mu}{el} = \sumI ZI \vec{R}_I - \int \rho(\vec{r}) \vec{r} d\vec{r} $$
where ZI and RI are the nuclear charges and coordinates, respectively [92]. The orbital energies obtained by diagonalizing the Fock matrix provide insights into molecular reactivity, with the energies of the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) being particularly significant.
Dipole Moment Calculation and Verification
Theoretical Framework
The molecular electric dipole moment serves as a key indicator of charge distribution, influencing intermolecular interactions, solvation behavior, and spectroscopic properties. For SCF methods, the dipole moment calculation follows the general formula in Equation 1, where the electronic component can be computed either through integration of the electron density or by summing dipole molecular orbital integrals for all occupied orbitals [92].
For ground-state systems, the dipole moment is computed directly from the converged SCF density. For excited states, ΔSCF methods offer an alternative approach by targeting non-Aufbau occupations, allowing excited-state properties to be computed with ground-state methodology [92]. However, careful attention must be paid to potential spin contamination in open-shell singlet states, which can affect the accuracy of the computed properties.
Accuracy Assessment and Benchmarking
Recent comprehensive benchmarks of excited-state dipole moments from ΔSCF methods reveal variable performance depending on the system and functional employed. On average, ΔSCF data do not necessarily improve upon time-dependent density functional theory (TDDFT) results but offer increased accuracy in specific cases, particularly for doubly excited states that are inaccessible to conventional TDDFT [92].
Table 1: Performance of Computational Methods for Dipole Moment Prediction
| Method | Strengths | Limitations | Typical RMSE/Relative Error |
|---|---|---|---|
| ΔSCF | Access to double excitations; Ground-state methodology for excited properties | Overdelocalization error in charge-transfer states; Spin contamination in open-shell singlets | Varies widely; can outperform TDDFT for specific cases [92] |
| TDDFT | Standard for single excitations; Broad availability | Fails for double excitations; Often overestimates dipole magnitude with standard functionals | ~28% (CAM-B3LYP) to ~60% (B3LYP, PBE0) for excited states [92] |
| Double Hybrid DFAs | High accuracy for ground states | Computational cost | ~4% regularized RMSE for ground states [92] |
| Hybrid DFAs | Balanced cost/accuracy | Systematic errors for certain excitations | ~6% regularized RMSE for ground states [92] |
| CCSD | High accuracy for excited states | High computational cost | ~10% average relative error for excited states [92] |
For ground-state equilibrium dipole moments, the performance of density functional approximations (DFAs) has been extensively benchmarked. Double hybrid functionals yield regularized root mean square errors (RMSE) of approximately 4%, comparable to coupled-cluster with single and double excitations (CCSD), followed by hybrid functionals (RMSE < 6%), and semilocal functionals (RMSE ≈ 8%) [92].
Experimental Protocols for Dipole Moment Calculation
Ground-State Dipole Moment Protocol:
- Perform SCF calculation with appropriate functional and basis set until full convergence
- Extract the converged density matrix or molecular orbitals
- Compute the electronic contribution to the dipole moment through integration of the electron density with the position operator
- Add the nuclear contribution based on molecular geometry
- For validation, compare with experimental measurements or high-level wavefunction theory
ΔSCF Excited-State Dipole Moment Protocol:
- Identify the target excited state and its character (singly excited, doubly excited, charge-transfer)
- Select appropriate ΔSCF variant (MOM, IMOM, σ-SCF, SGM, or STEP) to avoid variational collapse
- For open-shell singlet states, consider spin-purification techniques (e.g., ROKS) to improve accuracy
- Converge to the desired excited state determinant using appropriate convergence accelerators
- Compute dipole moment from the resulting electron density using standard ground-state formulae
- Validate against theoretical benchmarks or experimental data where available
Orbital Energy Analysis and Interpretation
Physical Significance of Orbital Energies
In SCF theories, the orbital energies obtained by diagonalizing the Fock matrix provide valuable insights into electronic structure and reactivity. The HOMO-LUMO gap offers a preliminary indicator of molecular stability and excitation properties, while the entire orbital energy spectrum can be used to interpret photoelectron spectra and understand chemical bonding patterns.
The orbital energies emerge from the generalized eigenvalue problem at the heart of SCF theory. In a non-orthogonal atomic orbital basis, this takes the form F C = S C E, where the columns of C contain the molecular orbital coefficients, and the diagonal matrix E contains the orbital energies [1] [20]. Transformation to an orthogonal basis simplifies this to a standard eigenvalue problem.
Methodological Considerations for Accurate Orbital Energies
The accuracy of orbital energies depends critically on the SCF convergence and the choice of exchange-correlation functional in DFT calculations. For difficult cases with small HOMO-LUMO gaps, techniques such as level shifting, fractional occupations, or smearing may be necessary to achieve convergence [1]. It is essential to perform stability analysis post-convergence to verify that the solution represents a true minimum rather than a saddle point [1].
Table 2: SCF Convergence Techniques for Orbital Energy Calculations
| Technique | Mechanism | Application Context | Key Parameters |
|---|---|---|---|
| DIIS | Extrapolates Fock matrix using previous iterations by minimizing commutator norm [1] | Standard acceleration method | DIIS space size; Start cycle |
| Damping | Mixes old and new Fock matrices to prevent oscillations [1] | Initial convergence phases; Oscillatory behavior | Damping factor (0.5-0.9) |
| Level Shifting | Increases gap between occupied and virtual orbitals [1] | Small HOMO-LUMO gaps; Near-degeneracies | Shift value (0.1-0.5 Eh) |
| SOSCF | Uses second-order orbital optimization for quadratic convergence [1] | High-accuracy requirements; Difficult cases | CIAH method |
| Fractional Occupations | Allows non-integer orbital occupations [1] | Metallic systems; Small-gap systems | Occupation smearing width |
Population Analysis Methods
Theoretical Background
Population analysis encompasses a family of techniques for partitioning the electron density among atoms, orbitals, or molecular fragments. These methods transform the delocalized molecular orbital description into a localized picture more aligned with chemical intuition. Though population analyses yield quantities that are not quantum mechanical observables, they provide valuable interpretive tools for understanding charge distributions and bonding patterns [100].
Mulliken Population Analysis
The Mulliken scheme partitions the total electron density using the assignment of basis functions to specific atoms and the overlap between them. The number of electrons on center A is calculated as:
$$ NA = \sum{\mu}^{A} \sum{\nu}^{A} P{\mu\nu}^{AA} S{\mu\nu}^{AA} + \sum{B \neq A} \sum{\mu}^{A} \sum{\nu}^{B} P{\mu\nu}^{AB} S{\mu\nu}^{AB} $$
where the first term represents the net population and the second term divides the overlap population equally between the two centers [100]. The atomic charge is then obtained as QA = ZA - NA, where ZA is the nuclear charge. The Mulliken bond order between atoms A and B is defined as:
$$ B{AB} = 2 \sum{\mu}^{A} \sum{\nu}^{B} P{\mu\nu}^{AB} S_{\mu\nu}^{AB} $$
Despite its simplicity and widespread implementation, the Mulliken analysis suffers from significant basis set dependence, with diffuse functions often leading to unphysical negative populations [100].
Löwdin and Mayer Population Analyses
The Löwdin analysis employs a symmetric orthogonalization procedure to transform the basis to one where all overlap integrals vanish. Using the Löwdin orthogonalization matrix S^(-1/2), the density matrix transforms as P^L = S^(1/2)PS^(1/2), and the atomic populations become:
$$ NA = \sum{\mu}^{A} P_{\mu\mu}^{L} $$
The bond order in the Löwdin scheme is defined via the Wiberg index:
$$ B{AB} = \sum{\mu}^{A} \sum{\nu}^{B} (P{\mu\nu}^{L})^2 $$
The Mayer analysis offers an alternative approach, with Mayer atomic charges being identical to Mulliken charges but with a different bond order definition that often provides better alignment with chemical intuition [100].
Practical Implementation and Protocol
Population Analysis Protocol:
- Perform converged SCF calculation with well-chosen basis set
- Avoid excessively diffuse basis functions that can cause Mulliken instability
- Extract density matrix and overlap matrix from calculation
- Apply selected population analysis method (Mulliken, Löwdin, or Mayer)
- Interpret results in context of chemical knowledge and compare multiple methods
Most quantum chemistry packages including ORCA and PySCF provide built-in functionality for population analyses. In ORCA, the !MULLIKEN and !LOEWDIN keywords trigger the respective analyses, with detailed output controllable through the %output block [100]. For frontier molecular orbital analysis, the !FMOPop keyword provides specialized population data for the HOMO and LUMO [100].
Table 3: Comparison of Population Analysis Methods
| Method | Basis | Charge Definition | Bond Order Definition | Advantages | Limitations |
|---|---|---|---|---|---|
| Mulliken | Original non-orthogonal | QA = ZA - NA with NA from gross population [100] | BAB = 2∑μ^A∑ν^B P{μν}^{AB}S_{μν}^{AB} [100] | Simple; Intuitive; Widely available | Strong basis set dependence; Can yield unphysical results |
| Löwdin | Orthogonalized (S^(-1/2)) | QA = ZA - NA with NA = ∑μ^A P{μμ}^L [100] | BAB = ∑μ^A∑ν^B (P{μν}^L)^2 [100] | More robust to basis set choice; Better bond orders | Less chemically intuitive transformation |
| Mayer | Original non-orthogonal | Same as Mulliken [100] | Based on quadratic density matrix products [100] | Improved bond orders for multiple bonds | Same charge limitations as Mulliken |
Visualization and Workflow Diagrams
SCF Property Calculation Workflow
Research Reagent Solutions: Computational Tools
Table 4: Essential Computational Tools for SCF Property Verification
| Tool Category | Specific Examples | Function in Property Verification |
|---|---|---|
| Quantum Chemistry Packages | PySCF [1], Q-Chem [2], ORCA [100], OpenMolcas [101] | Provide SCF solvers with various initial guesses, convergence accelerators, and property calculation capabilities |
| Initial Guess Methods | MINAO [1], Atom [1], Hückel [1], 1e [1], Checkpoint file [1] | Generate starting orbitals for SCF procedure to ensure convergence to desired state |
| Convergence Accelerators | DIIS [1], EDIIS [1], ADIIS [1], SOSCF [1], Damping [1], Level Shifting [1] | Stabilize SCF iterations for difficult cases with small gaps or near-degeneracies |
| Population Analysis Tools | Mulliken [100], Löwdin [100], Mayer [100], Frontier MO Populations [100] | Partition electron density to atomic centers and analyze bonding patterns |
| Stability Analysis | Internal/External Stability Check [1] | Verify that converged SCF solution represents true minimum rather than saddle point |
| Benchmarking Methods | ΔSCF [92], TDDFT [92], CCSD [92], CC2 [92] | Provide reference data for validation of predicted properties |
The verification of dipole moments, orbital energies, and population analyses within the SCF framework requires careful attention to methodological details and convergence criteria. Dipole moments serve as sensitive probes of electron distribution and can be computed for both ground and excited states using SCF and ΔSCF approaches, though their accuracy varies significantly with the choice of functional and system characteristics. Orbital energies provide valuable insights into electronic structure but require rigorous convergence and stability analysis. Population analyses transform the delocalized molecular orbital description into chemically intuitive concepts but must be interpreted with awareness of their limitations and methodological dependencies.
The continuing development of SCF methodologies, including improved initial guesses, convergence accelerators, and analysis techniques, enhances our ability to predict and verify molecular properties with increasing accuracy and reliability. For researchers in drug development and materials science, the rigorous application of the protocols and verification strategies outlined in this guide enables more confident utilization of computational predictions in experimental design and interpretation.
The self-consistent field (SCF) iterative process is a cornerstone of computational chemistry, underpinning quantum mechanical methods from Hartree-Fock to Kohn-Sham density functional theory (DFT). Its convergence to a stable electronic ground state is vital for predicting material properties, reaction mechanisms, and electronic structures. However, a fundamental tension persists between the accuracy of results and the computational cost required to achieve them. This analysis examines contemporary strategies—from advanced SCF algorithms to machine learning (ML)-driven approaches—that seek to redefine this trade-off. Framed within a broader guide to SCF process research, this review provides a quantitative and methodological comparison of these techniques, offering researchers a clear framework for selecting and implementing optimal computational strategies for drug development and materials science.
This section details the core methodologies, presenting their performance data in a structured format for direct comparison.
Machine Learning Potentials for Direct Property Prediction
Neural Network Potentials (NNPs) bypass the SCF process entirely by learning a direct mapping from atomic structure to system energy and properties.
- EMFF-2025: A general NNP for C, H, N, O-based high-energy materials. It uses a transfer learning strategy, building upon a pre-trained model with minimal DFT data to achieve high accuracy. Its performance is benchmarked against DFT calculations and experimental data [102].
- OMol25-Trained Models: These are pre-trained NNPs (eSEN and UMA architectures) on a massive dataset of ωB97M-V/def2-TZVPD calculations. They are designed for general molecular applications, including predicting charge-related properties like reduction potential and electron affinity [103].
Performance Benchmark Table: Machine Learning Potentials
| Method/Model | Target System | Accuracy Metric | Computational Cost | Key Limitation |
|---|---|---|---|---|
| EMFF-2025 [102] | Condensed-phase HEMs (C,H,N,O) | MAE: Energy < 0.1 eV/atom; Force < 2 eV/Å | DFT-level accuracy at MD simulation cost | Applicability limited to trained element types. |
| OMol25 (UMA-S) [103] | Main-group & organometallic molecules (Reduction Potential) | MAE: 0.261 V (OROP), 0.262 V (OMROP) | Lower than DFT/SQM | Accuracy varies with molecular set and property. |
| OMol25 (UMA-S) [103] | Main-group & organometallic molecules (Electron Affinity) | As accurate or better than low-cost DFT/SQM | Lower than DFT/SQM | Does not explicitly model charge/spin physics. |
Advanced Algorithms for SCF Convergence Acceleration
A critical path to efficiency is improving the SCF algorithm itself to reduce the number of iterations required for convergence.
- Adaptive Damping Algorithm: This parameter-free method replaces a fixed damping parameter with a backtracking line search based on an inexpensive energy model at each SCF step. It enhances robustness, especially in challenging systems like elongated supercells and transition-metal alloys [94].
- ADIIS (Augmented DIIS): This method combines Pulay's DIIS approach with the augmented Roothaan–Hall (ARH) energy function. It minimizes the ARH energy to obtain the linear coefficients for Fock matrix mixing, leading to more robust and efficient convergence than standard DIIS or EDIIS, particularly when combined with DIIS in an "ADIIS+DIIS" scheme [46].
- Bayesian Optimization of Mixing Parameters: This data-efficient algorithm systematically optimizes charge mixing parameters in DFT codes like VASP, reducing the number of SCF iterations needed for convergence without user intervention [104].
Performance Benchmark Table: SCF Algorithms & ML Accelerators
| Method/Model | Principle | Accuracy | Computational Efficiency | Key Limitation |
|---|---|---|---|---|
| Adaptive Damping [94] | Automatic, parameter-free line search | Maintains DFT accuracy | Robust convergence on challenging systems | - |
| ADIIS+DIIS [46] | Energy minimization via ARH function | Maintains DFT accuracy | More robust/efficient than DIIS or EDIIS | - |
| Bayesian Optimization [104] | Optimizes charge mixing parameters | Maintains DFT accuracy | Reduces SCF iterations | Requires initial data sampling. |
| DeePH + HONPAS [105] | ML-predicted Hamiltonian bypasses SCF | HSE06 hybrid functional accuracy | Enables >10,000 atom hybrid-DFT calculations | Depends on localized basis sets (NAO). |
| Electron Density Prediction [47] | ML predicts density in auxiliary basis | Enables SCF convergence from improved guess | 33.3% avg. SCF reduction; highly transferable | Requires auxiliary basis set. |
Machine Learning for SCF Initialization and Hamiltonian Prediction
Instead of replacing quantum mechanics, some methods use ML to generate high-quality initial guesses for the SCF cycle, drastically reducing iteration counts.
- DeepH + HONPAS: This method uses the DeepH framework to learn the mapping from atomic structure to the Hamiltonian matrix. When interfaced with HONPAS software, it bypasses the most time-consuming SCF iterations, enabling hybrid functional (HSE06) calculations for systems exceeding ten thousand atoms, a feat previously computationally intractable [105].
- Electron Density Prediction: A recently proposed paradigm trains an E(3)-equivariant neural network to predict the electron density represented in a compact auxiliary basis. This approach is shown to be more transferable and scalable than predicting the Hamiltonian or density matrix directly, achieving a significant reduction in SCF steps across different system sizes, basis sets, and functionals [47].
Experimental Protocols and Workflows
To ensure reproducibility and provide a clear guide for implementation, this section outlines detailed protocols for key methodologies cited in this analysis.
Protocol: Benchmarking NNPs on Redox Properties
This protocol, derived from benchmark studies on OMol25-trained models, details how to evaluate the accuracy of NNPs for predicting charge-related properties [103].
- Data Acquisition: Obtain a dataset containing molecular structures for both the non-reduced and reduced states of the species of interest, along with experimental reduction potential or electron affinity values.
- Geometry Optimization: Optimize the non-reduced and reduced structures using the target NNP. Tools like
geomeTRICcan be employed for this purpose. - Solvation Correction (for Reduction Potential): Input the optimized structures into a solvation model (e.g., CPCM-X) to obtain solvent-corrected electronic energies.
- Energy Difference Calculation: Calculate the predicted property.
- For reduction potential, compute: ( E{\text{non-red}} - E{\text{red}} ) (in eV), which gives the potential in volts.
- For electron affinity, compute: ( E{\text{neutral}} - E{\text{anion}} ) (in eV).
- Statistical Analysis: Compare the predicted values against experimental data using statistical metrics like Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), and the coefficient of determination (R²).
Protocol: Accelerating SCF via ML-Predicted Electron Density
This protocol describes the workflow for using a machine learning-predicted electron density to generate a high-quality initial guess for the SCF procedure [47].
- Model Training: Train an E(3)-equivariant neural network on a dataset of molecules (e.g., the SCFbench dataset) to predict the expansion coefficients ( ck ) of the electron density in a compact auxiliary basis set ( {\chik(\bm{r})} ).
- Density Construction: For a new molecule, use the trained model to predict the coefficients ( ck ). Construct the electron density as ( \rho(\bm{r}) \approx \sumk ck \chik(\bm{r}) ).
- Hamiltonian Assembly: Use the predicted density to directly compute the components of the Kohn-Sham Hamiltonian matrix.
- The Coulomb matrix (( \bm{J} )) is computed efficiently from the coefficients ( ck ) using the density fitting approximation.
- The exchange-correlation matrix (( \bm{V}{\text{xc}} )) is evaluated using the predicted density and its gradient (for GGA functionals).
- SCF Initialization: Initialize the SCF cycle with the Hamiltonian matrix constructed in Step 3. This provides a starting point much closer to the converged solution than standard guesses (e.g., superposition of atomic densities), significantly reducing the number of iterations required for convergence.
The following workflow diagram illustrates the core SCF process and the integration points for the major acceleration methods discussed.
This table catalogs key software, algorithms, and datasets that form the essential "reagents" for modern, efficient electronic structure research.
Research Reagent Solutions Table
| Item Name | Type | Function/Purpose | Key Reference |
|---|---|---|---|
| OMol25 Dataset | Dataset | Provides over 100 million computational chemistry calculations for pre-training and benchmarking general NNPs. | [103] |
| SCFbench Dataset | Dataset | A public benchmark for developing and testing DFT acceleration methods, focusing on electron density prediction. | [47] |
| EMFF-2025 | Neural Network Potential | A ready-to-use NNP for predicting mechanical and decomposition properties of high-energy materials at quantum accuracy. | [102] |
| DeepH Method | Software/Method | A machine learning framework for predicting DFT Hamiltonians, enabling large-scale hybrid functional calculations. | [105] |
| Adaptive Damping Algorithm | Algorithm | A parameter-free SCF convergence algorithm that automatically adjusts damping, increasing robustness. | [94] |
| ADIIS (ARH-DIIS) | Algorithm | An advanced SCF mixing scheme that uses energy minimization for robust convergence. | [46] |
| HONPAS | Software | A DFT package specializing in linear-scaling methods and efficient hybrid functional (HSE06) calculations for large systems. | [105] |
The landscape of electronic structure calculation is undergoing a rapid transformation driven by machine learning and sophisticated algorithms. The traditional trade-off between accuracy and computational cost is being actively redefined. For researchers, the choice of method depends on the specific problem: NNPs like EMFF-2025 offer unparalleled speed for property prediction within their trained domains, while advanced SCF algorithms and ML initializers like the electron-density-centric approach provide robust pathways to accelerate full quantum mechanical calculations for larger and more complex systems, including those relevant to drug development. The integration of these tools into a cohesive workflow, as illustrated in this analysis, promises to significantly accelerate the pace of scientific discovery.
Best Practices for Reproducible and Physically Meaningful SCF Results
The Self-Consistent Field (SCF) method represents a cornerstone computational procedure in electronic structure theory, forming the fundamental algorithm for Hartree-Fock and Density Functional Theory calculations across computational chemistry and materials science. The SCF procedure iteratively searches for a self-consistent electron density, where the computed quantum mechanical field and the resulting electron density become mutually consistent [50]. At each cycle, the electron density is computed from the occupied orbitals; this new density then defines the potential from which the orbitals are subsequently re-computed [69]. This cycle repeats until convergence is reached, signifying that the input and output densities are sufficiently similar according to a defined convergence criterion [50]. The core challenge lies in ensuring this iterative process converges reliably to a physically meaningful solution that represents the electronic ground state, rather than diverging or stalling in oscillatory behavior. This guide establishes comprehensive best practices for achieving reproducible, efficient, and physically meaningful SCF results, framed within the context of advancing reliable computational research, particularly in demanding applications like drug development where predictive accuracy is paramount.
Defining Convergence Criteria and Numerical Quality
Quantitative Convergence Thresholds
Convergence is typically assessed by the change in the density matrix or the commutator of the Fock and density matrices between iterations. Establishing appropriate convergence thresholds is critical for both numerical accuracy and computational efficiency. An excessively tight criterion wastes computational resources, while a loose criterion may yield insufficiently accurate results for subsequent property calculations.
Table 1: Standard SCF Convergence Criteria Across Major Quantum Chemistry Codes
| Software | Default Criterion | Key Parameter | Tight/Alternative Setting |
|---|---|---|---|
| ADF [69] | Commutator max element < 10⁻⁶ | SCF Converge |
Create mode default: 10⁻⁸ |
| BAND [50] | Density error < 10⁻⁶ × √Nₐₜₒₘₛ (Normal quality) | Convergence%Criterion |
VeryGood quality: 10⁻⁸ × √Nₐₜₒₘₛ |
| Q-Chem [49] | Wavefunction error < 10⁻⁵ (SP energy) / 10⁻⁸ (geometry opt, vibrations) | SCF_CONVERGENCE |
Must be compatible with integral threshold (THRESH) |
| Gaussian [15] | RMS density change < 10⁻ⁿ, Max change < 10⁻⁽ⁿ⁻²⁾ | SCF=Conver=N |
Typically requires ~10⁻²ᴺ energy change (atomic units) |
Integration with Numerical Grids and Precision
The SCF convergence criterion must be compatible with other numerical settings in the calculation. For instance, in Q-Chem, the integral threshold (THRESH) must be set at least three orders of magnitude tighter than the SCF_CONVERGENCE criterion [49]. Similarly, in Gaussian, using tighter SCF convergence (e.g., SCF=Tight) often coincides with finer integration grids (e.g., Int=Grid=UltraFine) to ensure consistent numerical accuracy across all components of the calculation [106]. Inefficient convergence or convergence to a physically meaningless solution can often be traced to inconsistencies between these numerical parameters.
SCF Convergence Algorithms and Acceleration Techniques
Algorithm Selection and Workflows
Choosing an appropriate convergence algorithm is the most critical step in ensuring robust SCF performance. Different algorithms offer varying trade-offs between stability, computational cost, and suitability for specific system types.
Table 2: SCF Convergence Algorithms and Their Applications
| Algorithm | Core Principle | Typical Use Case | Software Availability |
|---|---|---|---|
| DIIS (Pulay) [15] [49] | Extrapolates new Fock matrix from linear combination of previous iterations. | Default for most well-behaved, closed-shell systems. | Gaussian (default), Q-Chem, ADF |
| ADIIS + SDIIS [69] | Combines energy-directed (ADIIS) and residual-minimization (SDIIS) approaches. | Default in ADF's new SCF code; good for general purpose use. | ADF |
| QC (Quadratic Convergent) [15] | Uses second-order convergence methods (Newton-Raphson). | Difficult convergence cases; not available for ROHF. | Gaussian |
| GDM (Geometric Direct Minimization) [49] | Direct minimization of the energy with respect to orbital rotations. | Recommended for restricted open-shell calculations. | Q-Chem |
| LIST Family (LISTi, LISTb, LISTf) [50] [69] | Linear-expansion shooting techniques; sensitive to number of expansion vectors. | Alternative when DIIS fails; can be combined in MESA scheme. | ADF, BAND |
| S-GEK/RVO [107] | Uses gradient-enhanced Kriging surrogate model and restricted-variance optimization. | Emerging method for robust convergence and efficiency gains. | OpenMolcas (enhancements) |
Advanced Algorithmic Workflows and Hybrid Schemes
For particularly challenging systems, robust black-box workflows that combine multiple algorithms are recommended. Q-Chem's ROBUST and ROBUST_STABLE procedures exemplify this approach, featuring tighter thresholds and a strategic combination of algorithms (DIIS, ADIIS, and GDM) to maximize the probability of convergence [49]. Similarly, switching algorithms mid-calculation is a common strategy:
LS_DIIS: Uses level-shifting initially, then switches to DIIS [49].DIIS_GDM: Uses DIIS initially, then switches to geometric direct minimization [49].ADIIS_DIIS: Uses ADIIS initially, then switches to DIIS [49]. These hybrid methods leverage the rapid initial convergence of simpler algorithms and the stability of more robust methods near convergence.
Figure 1: Core SCF Iteration Logic and Algorithm Integration
Troubleshooting and Enhancing Problematic Convergence
Systematic Protocol for SCF Convergence Failure
When standard SCF procedures fail, a systematic approach to troubleshooting is essential. The following protocol outlines a step-by-step methodology for addressing convergence problems, progressing from simple to more advanced interventions.
Table 3: Systematic Troubleshooting Protocol for SCF Convergence Failure
| Step | Action | Key Parameters/Keywords | Expected Outcome & Rationale |
|---|---|---|---|
| 1. Initial Stabilization | Apply damping or Fermi smearing. | Mixing 0.1 (ADF) [69], SCF=Fermi (Gaussian) [15], ElectronicTemperature (BAND) [50]. |
Suppresses oscillation by reducing step size or fractional occupation. |
| 2. Algorithm Switch | Change core convergence algorithm. | SCF_ALGORITHM=RCA_DIIS or ADIIS_DIIS (Q-Chem) [49], AccelerationMethod=LISTb (ADF) [69]. |
Engages more robust optimization path. |
| 3. Refine Initial Guess | Improve starting density/guess. | InitialDensity=psi (BAND) [50], Guess=Always, SpinFlip for AFM states [50]. |
Provides better starting point closer to solution. |
| 4. Advanced Methods | Employ last-resort algorithms. | SCF=QC or SCF=XQC (Gaussian) [15], ROBUST_STABLE with analysis (Q-Chem) [49]. |
Quadratic convergence or full workflow with stability check. |
| 5. Geometric Inspection | Check for unrealistic molecular geometry. | --- | Eliminates non-physical starting points causing SCF failure. |
Special Considerations for Complex Systems
Specific categories of chemical systems present unique SCF convergence challenges:
- Metallic Systems and Small-Gap Semiconductors: These benefit from fractional occupation smearing (Fermi smearing or Gaussian broadening) to stabilize convergence around the Fermi level [50] [15].
- Open-Shell and Radical Systems: Using maximum spin initial guesses (
StartWithMaxSpin) and carefully handling spin flipping (SpinFlip,SpinFlipRegion) can help achieve the desired spin state and break initial symmetry [50]. - Transition Metal Complexes: These often require increased maximum SCF cycles (
Iterations,MAX_SCF_CYCLES) and may benefit from level shifting (Lshift) to prevent charge sloshing between near-degenerate metal and ligand orbitals [49] [69]. - Spin-Orbit Coupling Calculations: Specialized initial guess strategies (
StartWithMaxSpinForSO) are needed as level shifting is not supported in this context [50] [69].
Figure 2: Systematic SCF Troubleshooting Decision Tree
The Scientist's Toolkit: Essential Computational Reagents
Successful SCF calculations require the careful selection and combination of various "computational reagents" — the algorithms, parameters, and strategies that form the essential toolkit for modern computational researchers.
Table 4: Essential Research Reagent Solutions for SCF Calculations
| Tool Category | Specific Item/Parameter | Function/Purpose | Implementation Example |
|---|---|---|---|
| Core Solvers | DIIS, GDM, QC, LIST methods [15] [49] [69] | Extrapolate or minimize to find self-consistent solution. | SCF_ALGORITHM=GDM (Q-Chem) for open-shell. |
| Convergence Accelerators | Damping, Level Shifting, Fermi Smearing [15] [69] | Stabilize early iterations, prevent oscillation. | Mixing 0.2 (ADF), SCF=Fermi (Gaussian). |
| Initial Guess Handlers | Atomic density superposition, fragment guesses, saved orbitals [50] [15] | Provide physically reasonable starting point. | InitialDensity=psi (BAND), Guess=Read (Gaussian). |
| Spin & Symmetry Controllers | StartWithMaxSpin, SpinFlip, NoSymm [50] [15] |
Control initial spin state and break symmetry. | SpinFlip 1 3 flips spin on atoms 1 & 3 (BAND). |
| Numerical Thresholds | SCF convergence, integral cutoff, grid size [50] [49] [106] | Control numerical precision and accuracy. | SCF_CONVERGENCE 8, THRESH 14 (Q-Chem). |
Achieving reproducible and physically meaningful SCF results requires a systematic approach that integrates appropriate convergence criteria, carefully selected algorithms, and strategic troubleshooting protocols. The key principles outlined in this guide—understanding the relationship between numerical settings, selecting algorithms matched to specific system characteristics, and following a structured response to convergence failures—provide a foundation for reliable computational research. As SCF methodologies continue to evolve with emerging approaches like S-GEK/RVO [107] and robust hybrid algorithms [49], adherence to these best practices will ensure that computational results remain both reproducible and physically interpretable, thereby strengthening the bridge between theoretical computation and experimental science in critical fields like drug discovery and materials design.
Conclusion
The SCF iterative process remains fundamental to computational chemistry and materials science, with proper implementation crucial for reliable results in drug discovery and biomedical research. Mastering both theoretical foundations and practical troubleshooting techniques enables researchers to efficiently handle diverse molecular systems. Future directions include increased integration of machine learning for Hamiltonian prediction and initial guesses, development of more robust convergence algorithms for complex biomolecules, and enhanced validation protocols to ensure physical meaningfulness of solutions. As computational demands grow in pharmaceutical research, understanding and optimizing the SCF process will continue to be essential for accelerating discovery and innovation.
References
- 1. Self-consistent field (SCF) methods [https://pyscf.org/user/scf.html]
- 2. 4.1 Overview‣ Chapter 4 Self-Consistent Field ... [https://manual.q-chem.com/5.3/Ch4.S1.html]
- 3. Timeline: The continuing evolution of density functional ... [https://www.microsoft.com/en-us/research/articles/timeline-the-continuing-evolution-of-density-functional-theory/]
- 4. Hartree–Fock method [https://en.wikipedia.org/wiki/Hartree%E2%80%93Fock_method]
- 5. 4.2.2 Hartree-Fock Theory [https://manual.q-chem.com/6.3/Ch4.S2.SS2.html]
- 6. 4.5 Converging SCF Calculations [https://manual.q-chem.com/5.1/sect-convergence.html]
- 7. SCF Convergence Guidelines for ADF - SCM [https://www.scm.com/doc/ADF/Rec_problems_questions/SCF.html]
- 8. The self-consistent-field cycle - The SIESTA project [https://docs.siesta-project.org/projects/siesta/en/5.0/tutorials/basic/scf-convergence/]
- 9. 7.7. SCF Convergence - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/scfconv.html]
- 10. Kohn-Sham Density-Functional Theory - Mathematical Sciences [https://math.rpi.edu/events/kohn-sham-density-functional-theory-perspectives-various-inverse-problems]
- 11. 2. Fock matrices and the SCF procedure [https://kthpanor.github.io/cb2070/docs/exercise-fock.html]
- 12. The DePrince Research Group [https://www.chem.fsu.edu/~deprince/programming_projects/scf/index.php]
- 13. HF: Hartree–Fock Theory [https://psicode.org/psi4manual/1.9.x/scf.html]
- 14. *SCF — DIRAC 21.1 documentation [https://www.diracprogram.org/doc/release-21/manual/wave_function/scf.html]
- 15. SCF [https://gaussian.com/scf/]
- 16. 4.6.4 Incremental and Variable Thresh Fock Matrix Building [https://manual.q-chem.com/5.3/sect_incfock.html]
- 17. Linear combination of atomic orbitals [https://en.wikipedia.org/wiki/Linear_combination_of_atomic_orbitals]
- 18. Basis set (chemistry) [https://en.wikipedia.org/wiki/Basis_set_(chemistry)]
- 19. 18.5: The LCAO-MO Expansion [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Quantum_Mechanics__in_Chemistry_(Simons_and_Nichols)/18%3A_Multiconfiguration_Wavefunctions/18.05%3A_The_LCAO-MO_Expansion]
- 20. An Overview of Self-Consistent Field Calculations Within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7179435/]
- 21. 4.2.1 SCF and LCAO Approximations [https://manual.q-chem.com/6.2/Ch4.S2.SS1.html]
- 22. Self-consistent field methods - DFTK.jl [https://docs.dftk.org/stable/guide/self_consistent_field/]
- 23. Acceleration of Self-Consistent Field Calculations Using ... [https://pubmed.ncbi.nlm.nih.gov/39876602/]
- 24. 18.4: Atomic Orbital Basis Sets [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Quantum_Mechanics__in_Chemistry_(Simons_and_Nichols)/18%3A_Multiconfiguration_Wavefunctions/18.04%3A_Atomic_Orbital_Basis_Sets]
- 25. Correlation-consistent Gaussian basis sets for solids made ... [https://arxiv.org/abs/2112.05824]
- 26. Mean-field theory [https://en.wikipedia.org/wiki/Mean-field_theory]
- 27. Electronic Structure Calculations using Dynamical Mean ... [https://arxiv.org/abs/cond-mat/0511293]
- 28. Dynamical mean-field theory [https://en.wikipedia.org/wiki/Dynamical_mean-field_theory]
- 29. The self-consistent-field cycle - The SIESTA project [https://docs.siesta-project.org/projects/siesta/en/latest/tutorials/basic/scf-convergence/]
- 30. Roothaan-Hall equations [https://www.schulz.chemie.uni-rostock.de/storages/uni-rostock/Alle_MNF/Chemie_Schulz/Computerchemie_2/ro-hall.html]
- 31. Roothaan equations - Wikipedia [https://en.wikipedia.org/wiki/Roothaan_equations]
- 32. Unrestricted Hartree–Fock [https://en.wikipedia.org/wiki/Unrestricted_Hartree%E2%80%93Fock]
- 33. The Roothaan-Hall self-consistent field procedure [https://enccs.github.io/veloxchem-workshop/notebooks/rh-scf/]
- 34. 9.7: The Self-Consistent Field Approximation (Hartree-Fock ... [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Book%3A_Quantum_States_of_Atoms_and_Molecules_(Zielinksi_et_al)/09%3A_The_Electronic_States_of_the_Multielectron_Atoms/9.07%3A_The_Self-Consistent_Field_Approximation_(Hartree-Fock_Method)]
- 35. 4.2 Hartree–Fock Calculations [https://manual.q-chem.com/4.3/sect0037.html]
- 36. Basic Roothaan-Hall Equations [http://openmopac.net/manual/Roothaan_Hall.html]
- 37. Self-Consistent Field Iteration [https://www.emergentmind.com/topics/self-consistent-field-iteration]
- 38. Self-consistent basis set extrapolation of Hartree–Fock ... [https://link.springer.com/article/10.1007/s11224-025-02542-2]
- 39. Learning the One-Electron Reduced Density Matrix at SCF ... [https://chemrxiv.org/engage/chemrxiv/article-details/68c9a9763e708a76498380b5]
- 40. 4.4 SCF Initial Guess [https://manual.q-chem.com/4.3/sect-initialguess.html]
- 41. 4.4.2 Initial Guess Types [https://manual.q-chem.com/5.4/sect_guess.html]
- 42. Guess [https://gaussian.com/guess/]
- 43. Atomic start guess [https://www.diracprogram.org/doc/release-16/tutorials/start_guess/atomic_start.html]
- 44. Semiempirical Quantum Chemistry in the Age of ab initio ... [https://chemrxiv.org/engage/chemrxiv/article-details/68b45169728bf9025e6d3cfb]
- 45. 4.5.3 Direct Inversion in the Iterative Subspace (DIIS) [https://manual.q-chem.com/6.2/sect_diis.html]
- 46. Accelerating self-consistent field convergence with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2830258/]
- 47. Towards A Universally Transferable Acceleration Method ... [https://arxiv.org/html/2509.25724v1]
- 48. Difficult cases for converging Kohn-Sham SCF calculations [https://mattermodeling.stackexchange.com/questions/495/difficult-cases-for-converging-kohn-sham-scf-calculations]
- 49. 4.5.2 Basic Convergence Control Options [https://manual.q-chem.com/6.3/Ch4.S5.SS2.html]
- 50. Self Consistent Field (SCF) — BAND 2025.1 documentation [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/SCF.html]
- 51. Open-Source Projects We Wish Existed - Rowan Scientific [https://rowansci.com/blog/open-source-projects-we-wish-existed]
- 52. — BAND Basis .1 documentation set 2025 [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/Basis_Set.html]
- 53. Impact of the Intramolecular Basis Set Superposition Error [https://pmc.ncbi.nlm.nih.gov/articles/PMC6832644/]
- 54. What considerations must be made when selecting a basis ... [https://mattermodeling.stackexchange.com/questions/13987/what-considerations-must-be-made-when-selecting-a-basis-set]
- 55. issue with diffuse functions on SCF - Questions convergence basis set [https://talk.q-chem.com/t/scf-convergence-issue-with-diffuse-functions-on-basis-set/2925]
- 56. Could Someone Give me Advice on Optimizing Basis Set ... [https://forum.psicode.org/t/could-someone-give-me-advice-on-optimizing-basis-set-selection-for-calculating-reaction-energies/3108]
- 57. SCF Convergence Issues - ORCA Input Library [https://sites.google.com/site/orcainputlibrary/scf-convergence-issues]
- 58. SCF convergence problem with large basis sets [https://groups.google.com/g/cp2k/c/6JDb5uy5r0M]
- 59. Multi-configurational self-consistent field [https://en.wikipedia.org/wiki/Multi-configurational_self-consistent_field]
- 60. Multi-configuration self-consistent field (MCSCF) [https://pyscf.org/user/mcscf.html]
- 61. Quantum techniques offer a glimpse of next-generation ... [https://www.drugdiscoverynews.com/quantum-techniques-offer-a-glimpse-of-next-generation-medicine-16815]
- 62. The SCF program [https://www.molpro.net/manual/doku.php?id=the_scf_program]
- 63. Density matrix renormalization group [https://en.wikipedia.org/wiki/Density_matrix_renormalization_group]
- 64. Quantum computing in life sciences and drug discovery [https://www.mckinsey.com/industries/life-sciences/our-insights/the-quantum-revolution-in-pharma-faster-smarter-and-more-precise]
- 65. Iterative diagonalization in augmented plane wave based ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999109005282]
- 66. ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/casscf.html]
- 67. Weighted Active Space Protocol for Multireference ... [https://arxiv.org/html/2505.10505v1]
- 68. The complete active space SCF (CASSCF) method in a ... [http://ui.adsabs.harvard.edu/abs/1981JChPh..74.2384S/abstract]
- 69. SCF — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/SCF.html]
- 70. SCF Convergence and Chaos Theory [https://www.sci.muni.cz/~physics/CC/converge.htm]
- 71. A density matrix approach to the convergence of the self- ... [https://www.aimsciences.org/article/doi/10.3934/naco.2020018]
- 72. What are the physical reasons if the SCF doesn't converge? [https://mattermodeling.stackexchange.com/questions/8635/what-are-the-physical-reasons-if-the-scf-doesnt-converge]
- 73. Convergence Questions [https://downloads.wavefun.com/FAQ/converge.html]
- 74. 7.6. Choice of Initial Guess and Restart of SCF Calculations [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/initguess.html]
- 75. Optimization of Grand-Canonical Self-Consistent-Field ... [https://chemrxiv.org/engage/chemrxiv/article-details/68e225835dd091524ff23629]
- 76. 2.6. Self-Consistent-Field (SCF) [https://orca-manual.mpi-muelheim.mpg.de/contents/essentialelements/scf.html]
- 77. 4.5.4 Damping‣ 4.5 Converging SCF Calculations ‣ ... [https://manual.q-chem.com/5.3/sect_damp.html]
- 78. OpenOrbitalOptimizer—a reusable open source library for ... [https://arxiv.org/html/2503.23034v1]
- 79. The self-consistent-field cycle - The SIESTA project [https://docs.siesta-project.org/projects/siesta/en/stable/tutorials/basic/scf-convergence/]
- 80. 4.5.3 Direct Inversion in the Iterative Subspace (DIIS) [https://manual.q-chem.com/5.4/sect_diis.html]
- 81. 4.5.10 Small-Gap Systems [https://manual.q-chem.com/5.2/Ch4.S5.SS10.html]
- 82. A review on the derivation of the spin-Restricted Hartree— ... [https://www.sciencedirect.com/science/article/pii/S0010465598001003]
- 83. Methods to Solve the SCF not Converged | Zhe WANG, Ph.D. [https://wongzit.github.io/method-to-solve-the-scf-not-converged/]
- 84. Occupation Methods - QuantumATK [https://docs.quantumatk.com/manual/technicalnotes/occupation_methods/occupation_methods.html]
- 85. What do we physically mean by smearing in condensed ... [https://physics.stackexchange.com/questions/360037/what-do-we-physically-mean-by-smearing-in-condensed-matter]
- 86. How do you show that thermal broadening at Fermi ... [https://physics.stackexchange.com/questions/635415/how-do-you-show-that-thermal-broadening-at-fermi-distribution-is3-5kt-when-the]
- 87. Fermi liquid theory [https://en.wikipedia.org/wiki/Fermi_liquid_theory]
- 88. Approximate second order method for orbital optimization ... [https://link.springer.com/article/10.1007/s002140050241]
- 89. 7.11. SCF Stability Analysis - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/stabilityanalysis.html]
- 90. ∆SCF Excited-State Approach: Theoretical Foundation ... [https://archive.aps.org/smt/2025/mar-l67/9/]
- 91. [PDF] Saddle Point Search Algorithms for Variational ... [https://www.semanticscholar.org/paper/Saddle-Point-Search-Algorithms-for-Variational-of-Schmerwitz-Oll%C3%A9/65a62e0057dc74a1af5e50a60a078d694e3b80f5]
- 92. Excited state dipole moments from ΔSCF: a benchmark [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01695a]
- 93. Excited state (geometry) optimizations — ADF 2025.1 ... - SCM [https://www.scm.com/doc/ADF/Input/Excited_state_optimizations.html]
- 94. A robust and efficient line search for self-consistent field ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999122001899]
- 95. 4.5.9 Internal Stability Analysis and Automated Correction ... [https://manual.q-chem.com/5.2/Ch4.S5.SS9.html]
- 96. Wave Function Theory - an overview [https://www.sciencedirect.com/topics/chemistry/wave-function-theory]
- 97. 4.2.1 SCF and LCAO Approximations [https://manual.q-chem.com/5.2/Ch4.S2.SS1.html]
- 98. Projects: Organization, Sharing, and Saving Structures [https://rowansci.substack.com/p/projects-organization-sharing-and]
- 99. CHAOS - A Consistent Large-scale Database for σ [https://arxiv.org/html/2511.19002v1]
- 100. 5.1. Population Analysis [https://orca-manual.mpi-muelheim.mpg.de/contents/spectroscopyproperties/population.html]
- 101. A Self-Consistent Field program and Kohn–Sham DFT [https://molcas.gitlab.io/OpenMolcas/sphinx/tutorials/tut_scf.html]
- 102. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 103. Benchmarking OMol25-Trained Models on Experimental ... [https://rowansci.com/publications/omol25-redox]
- 104. Simple approach to more efficient density functional theory ... [https://www.sciencedirect.com/science/article/abs/pii/S2352214325000115]
- 105. Combining DeepH with HONPAS for accurate and efficient ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00128e]
- 106. Gaussian: Benchmark of G16 on Intel Xeon Gold 6148 [https://sites.google.com/site/rangsiman1993/comp-chem/test-and-benchmarks/gaussian-benchmark-g16-gold-6148]
- 107. Accelerating SCF Orbital Optimization with S-GEK/RVO [https://chemrxiv.org/engage/chemrxiv/article-details/68ecfc325dd091524f3a5b28]