Ionic Migration Dynamics on Perovskite Quantum Dot Surfaces: Mechanisms, Control Strategies, and Biomedical Applications
This article comprehensively explores the dynamics of ionic migration across the surfaces of perovskite quantum dots (PQDs), a critical factor influencing their stability and functionality.
Ionic Migration Dynamics on Perovskite Quantum Dot Surfaces: Mechanisms, Control Strategies, and Biomedical Applications
Abstract
This article comprehensively explores the dynamics of ionic migration across the surfaces of perovskite quantum dots (PQDs), a critical factor influencing their stability and functionality. Tailored for researchers and drug development professionals, it delves into the fundamental principles driving ion movement, advanced characterization and control methodologies, strategies for troubleshooting performance loss, and comparative analyses of effectiveness for biomedical applications. By synthesizing the latest research, this review establishes a framework for harnessing ionic migration to engineer next-generation PQD-based platforms for drug delivery, biosensing, and diagnostic imaging, bridging materials science with clinical translation.
Unraveling the Fundamentals: What Drives Ionic Migration on PQD Surfaces?
Defining Ionic Migration in the Soft Lattice of Halide Perovskites
Ionic migration refers to the movement of ions within the crystal lattice of a material under the influence of external stimuli such as an electric field, light, or heat. In halide perovskites, this phenomenon is particularly pronounced due to their intrinsic soft ionic lattice and mixed ionic-electronic conduction properties [1] [2]. The soft lattice, characterized by weak bonding and low formation energies for defects, allows for significant ionic movement, which dominates many of the anomalous phenomena observed in perovskite devices, including hysteresis, phase segregation, and slow response times [3]. Understanding and quantifying this ion migration is therefore a critical research gap that must be addressed to stabilize perovskite devices and enable their widespread commercialization [4] [1].
This guide provides an in-depth technical examination of ionic migration, framing the discussion within the broader context of ionic migration dynamics in perovskite quantum dot (PQD) surfaces research. It details the fundamental mechanisms, quantitative assessment techniques, and experimental protocols essential for researchers and scientists working to control ionic activity for enhanced device performance and longevity.
Fundamental Mechanisms and Theoretical Frameworks
Ionic migration in halide perovskites is a solid-state electrochemical phenomenon wherein vacancies and interstitial defects facilitate the hopping of ions through the crystal lattice [2]. The primary mobile species are typically halide anions (e.g., I⁻, Br⁻) and A-site cations (e.g., MA⁺, FA⁺), although the specific mobility depends on the composition and structure of the perovskite [1].
The Role of the Soft Lattice
The "soft" nature of the halide perovskite lattice, with its low elastic moduli and weak chemical bonding, results in low activation energies for defect migration. This structural softness is a double-edged sword: it contributes to the remarkable optoelectronic properties of perovskites but also makes them inherently prone to ionic movement, even under mild external biases [1] [2]. This movement can lead to deleterious effects such as:
- Chemical degradation: Ionic migration can facilitate reactions with electrode materials or environmental species [4].
- Phase segregation: In mixed-halide perovskites, ion migration can lead to the formation of halide-rich domains, undermining optoelectronic performance [3].
- Electronic band distortion: The accumulation of ions at interfaces and grain boundaries creates strong local electric fields that can distort the electronic band structure [2].
Table 1: Key Ion Migration Measurement Techniques and Representative Data
| Technique | Measured Parameter | Representative Value | Material System | Conditions |
|---|---|---|---|---|
| Non-contact PL Microscopy [2] | Ion Mobility | (2.56 ± 0.67) × 10⁻¹⁰ cm² V⁻¹ s⁻¹ | Single-crystal MAPbBr₃ | Room Temperature, Non-contact |
| Light-Enhanced Transport [3] | Activation Energy (Eₐ) | 0.82 eV (dark) → 0.15 eV (light, 20 mW/cm²) | MAPbI₃ Thin Film | 17-295 K, Under Illumination |
| High-Field Poling [3] | Ionic Conductance | Obtained via cryogenic galvanostatic measurement | MAPbI₃ | Lateral Au/MAPbI₃/Au Structure |
Theoretical and Computational Toolsets
Theoretical modeling plays a crucial role in understanding ion migration. Density Functional Theory (DFT) calculations, particularly with the Nudged Elastic Band (NEB) method, are widely used to calculate migration barriers (Eₘ), which correspond to the energy required for an ion to hop between two stable lattice sites [5] [1]. However, DFT-NEB can be computationally expensive and may underestimate Eₘ by 0.1–0.3 eV [5].
Empirical force fields like the Bond Valence Site Energy (BVSE) method offer a faster alternative for calculating percolation barriers, which represent the minimal energy barrier for infinite diffusion through the crystal, showing good correlation with DFT and experimental results [5]. Recently, Machine Learning Interatomic Potentials (MLIPs) have emerged as a powerful tool, enabling large-scale molecular dynamics simulations with near-DFT accuracy but at a fraction of the computational cost [5] [6]. Universal MLIPs like CHGNet and M3GNet are trained on diverse chemical spaces and can be fine-tuned for specific tasks, such as modeling the complex, charge-coupled dynamics of transition metal migration in materials like Mn-rich disordered rocksalt cathodes [6].
Quantitative Assessment and Data Presentation
Accurately quantifying ion migration is essential for understanding its impact and developing mitigation strategies. The following data, consolidated from recent studies, provides key quantitative benchmarks for the field.
Table 2: Computational Methods for Predicting Ionic Migration
| Computational Method | Key Parameter | Typical Use Case | Advantages | Limitations |
|---|---|---|---|---|
| DFT-NEB [5] | Migration Barrier (Eₘ) | Precise hop barrier calculation | High accuracy for specific pathways | Computationally expensive; underestimates Eₘ by 0.1-0.3 eV |
| BVSE [5] | Percolation Barrier (Eₐ) | High-throughput screening | Fast; good correlation with DFT/experiment | Empirical force field; less precise |
| AIMD [5] | Ionic Conductivity | Direct evaluation of ionic mobility | Captures lattice dynamics & correlation effects | Extremely computationally demanding |
| uMLIP (e.g., CHGNet) [5] [6] | Migration Trajectory & Barrier | Large-scale MD simulations of complex processes | Near-DFT accuracy, lower cost | Requires fine-tuning for specific chemistries |
Experimental Protocols for Probing Ionic Migration
Non-Contact Measurement of Ion Mobility via PL Microscopy
This protocol details a method to measure intrinsic ion mobility in single perovskite particles without direct electrical contact, thereby decoupling interfacial effects from bulk ion migration [2].
- Objective: To obtain the intrinsic ion mobility of single MAPbBr₃ particles in a non-contact manner.
- Materials and Reagents:
- Interpenetrating Electrode Device: Fabricated on a quartz substrate with a 10 μm gap [2].
- MAPbBr₃ Perovskite Particles: Synthesized and spin-coated onto the device.
- PL Microscopy System: Equipped with a high-sensitivity camera and appropriate laser excitation.
- Function Generator & Voltage Amplifier: To apply a modulated electric field (e.g., square wave, 130 kV/cm).
- Data Acquisition Card: To synchronize voltage application and PL signal recording.
- Procedure:
- Sample Preparation: Spin-coat a diluted MAPbBr₃ solution onto the interpenetrating electrode device to grow isolated single particles in situ. Ensure the studied particles are on the bare quartz substrate in the middle of the electrode gap, not in direct contact with the electrodes [2].
- Electric Field Application: Apply a periodic square-wave electric field (e.g., 130 kV/cm, 30-second period) across the electrodes using the function generator and voltage amplifier.
- PL Image Acquisition: Simultaneously record the PL intensity of the individual particles under the modulated electric field. The acquisition should be synchronized with the voltage signal.
- Data Analysis:
- Plot the evolution of PL intensity versus time.
- Calculate the Switching Efficiency (SE) using the formula: ( SE = (I{OFF} - I{ON}) / I{OFF} ), where ( I{OFF} ) and ( I_{ON} ) are the PL intensities with the field off and on, respectively [2].
- Analyze the polarization and recovery dynamics (time constants) from the PL intensity trajectory.
- Correlate the dynamics with particle size and electric field strength to calculate intrinsic ion mobility, which was found to be ((2.56 ± 0.67) \times 10^{-10} \, \text{cm}^2 \, \text{V}^{-1} \, \text{s}^{-1}) for MAPbBr₃ [2].
- Key Considerations: The non-contact setup is crucial for avoiding carrier injection and interfacial charge transport effects. The slow, second-scale PL dynamics are attributed to ion migration, as opposed to nanosecond-scale electronic processes [2].
Quantifying Light-Enhanced Ionic Transport
This protocol measures the influence of light illumination on ionic transport parameters, such as activation energy, across a wide temperature range [3].
- Objective: To quantitatively demonstrate the reduction of ionic transport activation energy under photoexcitation.
- Materials and Reagents:
- Lateral Device Structure: A patterned substrate (e.g., glass) with Au electrodes (e.g., 50 μm gap).
- MAPbI₃ Perovskite Film: Deposited onto the substrate covering the electrode gap.
- Cryogenic Station: Capable of maintaining temperatures from 17 K to room temperature.
- Precision Source/Measure Unit: For current-voltage (I-V) and galvanostatic measurements.
- Light Source: A fiber optic illuminator with adjustable intensity (0-20 mW cm⁻²).
- Procedure:
- Device Fabrication: Fabricate a lateral Au/MAPbI₃/Au device using a hard mask or lithography. Mount the device in the cryogenic station.
- Cryogenic Measurement: For a set of temperatures (e.g., from 17 K to 295 K), perform cryogenic galvanostatic and I-V measurements under different light intensities (0, 0.05, 1, 5, and 20 mW cm⁻²) [3].
- Conductance Separation: At each temperature and light intensity, separate the total measured conductance into its electronic and ionic components. This can be achieved by analyzing the transient response in galvanostatic measurements, where the instantaneous jump corresponds to electronic conductance and the slow relaxation corresponds to ionic conductance [3].
- Activation Energy Calculation:
- Plot the extracted pure ionic conductance as an Arrhenius plot (log of conductance vs. 1/T) for each light intensity.
- Fit the data to the Arrhenius equation. The slope of the fit is proportional to the activation energy (Eₐ) for ionic transport.
- Expected Outcome: A significant reduction in activation energy with increasing light intensity. For MAPbI₃, Eₐ decreases from ~0.82 eV in the dark to ~0.15 eV under 20 mW cm⁻² illumination [3].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Ion Migration Studies
| Item | Function/Application | Example Use Case |
|---|---|---|
| Interpenetrating Electrode Device [2] | Creates a non-contact electric field to polarize samples for intrinsic property measurement. | Non-contact PL measurement of ion mobility in single particles. |
| Lateral Au/MAPbI₃/Au Device [3] | Standard platform for applying in-plane electric fields and measuring lateral transport. | High-field poling experiments and cryogenic I-V measurements. |
| Methylammonium Halide (MAI) [3] | Organic precursor for synthesizing hybrid organic-inorganic halide perovskites (e.g., MAPbI₃, MAPbBr₃). | Sample fabrication for ion migration studies. |
| Lead(II) Iodide (PbI₂) [3] | Inorganic precursor for perovskite synthesis. | Used in sequential deposition methods for perovskite formation. |
| Bond Valence Site Energy (BVSE) Model [5] | Empirical force field for rapid calculation of ion percolation barriers. | High-throughput computational screening of ionic conductors. |
| Universal ML Interatomic Potentials (uMLIPs) [5] [6] | Machine-learned potentials for large-scale molecular dynamics with near-DFT accuracy. | Modeling complex phase transformations and ion migration trajectories. |
Ionic migration in the soft lattice of halide perovskites is a defining characteristic that underpins both the promise and the challenges of this material class. A comprehensive understanding, achieved through the synergistic application of advanced experimental techniques like non-contact PL microscopy and robust computational tools like machine learning interatomic potentials, is paramount. The precise quantification of parameters such as ion mobility and activation energy, and the development of strategies to control ionic movement, are critical steps toward overcoming stability issues and unlocking the full potential of halide perovskites in next-generation optoelectronic devices.
Metal halide perovskite quantum dots (PQDs), with the general formula ABX₃ (where A is a cation, B is a metal cation, and X is a halide anion), have emerged as a revolutionary class of semiconductor nanomaterials for optoelectronic applications. Their exceptional properties—including high photoluminescence quantum yield (PLQY), tunable bandgaps, and defect tolerance—are intrinsically linked to their ionic crystal nature [7] [8]. However, this ionic character is a double-edged sword, as it facilitates ion migration within the perovskite lattice, which is a primary factor influencing both the performance and operational stability of PQD-based devices [8]. The dynamics of halide anions (I⁻, Br⁻), A-site cations (Cs⁺, FA⁺), and B-site metal ions (Pb²⁺) at surfaces and interfaces dictate critical processes such as phase segregation, non-radiative recombination, and structural degradation [9] [8]. This whitepaper provides an in-depth technical analysis of the roles and migration behaviors of these key mobile ions, framing the discussion within the broader research context of controlling ionic dynamics to enhance the functional longevity of PQD technologies.
The Roles and Dynamics of Halide Anions (I⁻, Br⁻)
Structural and Optoelectronic Functions
Halide anions (X-site) form [BX₆]⁴⁻ octahedra, the fundamental building blocks of the perovskite crystal structure. The identity of the halide directly controls the material's bandgap and, consequently, its optical emission and absorption characteristics. Mixed halide perovskites (e.g., CsPbIₓBr₃₋ₓ) allow for continuous bandgap tuning across the visible spectrum [10] [11]. Despite their crucial role, halide ions are highly mobile due to their relatively low migration energy barriers, which makes them the most prevalent migratory species in the perovskite lattice [8].
Migration Mechanisms and Consequences
The primary mechanism for halide migration is believed to be vacancy-assisted diffusion [8]. The low formation energy of halide vacancies (Vₓ) facilitates their creation, providing pathways for adjacent halide ions to hop into these vacant sites. This process is accelerated by external stimuli such as electric fields, light, and heat [10].
Ion migration, particularly in mixed-halide systems, leads to several detrimental phenomena:
- Phase Segregation: Under illumination or electrical bias, halides can demix, leading to the formation of I-rich and Br-rich domains. This segregation causes undesirable changes in the emission spectrum and open-circuit voltage losses in solar cells [10].
- Non-Radiative Recombination: The movement of halide ions can create and mobilize deep-level traps within the bandgap, which act as centers for non-radiative recombination, thereby reducing PLQY and device efficiency [8] [11].
- Structural Instability: Progressive halide migration can initiate the decomposition of the perovskite crystal structure, ultimately leading to the formation of PbI₂ or other degradation products [9].
Table 1: Characteristics and Migration Behaviors of Key Halide Anions
| Halide Ion | Ionic Radius (Å) | Key Role | Migration Energy (eV) | Primary Instability Consequence |
|---|---|---|---|---|
| Iodide (I⁻) | ~2.20 | Red-shifted emission, narrow bandgap [10] | Low (est. < 0.1-0.5) | Phase segregation, photo-induced halide migration [10] |
| Bromide (Br⁻) | ~1.96 | Green emission, moderate bandgap [10] | Low (est. < 0.1-0.5) | Phase segregation in mixed halides, defect formation [8] |
The Roles and Dynamics of A-Site Cations (Cs⁺, FA⁺)
Structural Stabilization and Electronic Effects
A-site cations, situated in the cuboctahedral cavities of the [BX₆]⁴⁻ framework, are crucial for stabilizing the perovskite crystal structure. The Goldschmidt tolerance factor (t), which depends on the ionic radii of the A, B, and X ions, is a key predictor of structural stability [11]. While traditionally considered spatially confined, A-site cations, particularly smaller ions like Cs⁺, exhibit a degree of mobility that can significantly impact material properties [7] [9].
- Cesium (Cs⁺): The small ionic radius of the all-inorganic Cs⁺ cation helps stabilize the perovskite lattice against moisture-induced degradation compared to organic cations. However, Cs-rich PQDs are prone to thermally-induced phase transitions from the black γ-phase to a non-perovskite yellow δ-phase [9].
- Formamidinium (FA⁺): The larger organic FA⁺ cation contributes to a more ideal tolerance factor, often resulting in a narrower bandgap and enhanced orbital overlap, which leads to longer carrier lifetimes [7]. FA-rich PQDs exhibit stronger electron–longitudinal optical (LO) phonon coupling, which can dissociate photogenerated excitons [9].
Cation Exchange as a Migration Pathway
A prominent manifestation of A-site cation mobility is the post-synthetic cation-exchange process [7]. This reaction is driven by concentration gradients and a dynamic surface structure, allowing for the synthesis of mixed A-site (e.g., Cs₁₋ₘFAₘPbI₃) PQDs with tailored properties. The kinetics of this exchange are influenced by the surrounding chemical environment, including cation species, stoichiometric ratios, and surface ligand conditions [7]. The exchange process is theorized to proceed via a cation vacancy-assisted model, where vacancies on the A-site facilitate the exchange of cations between the solution and the crystal lattice [7].
Table 2: Comparison of A-Site Cations in PQDs
| A-Site Cation | Ionic Radius (Å) | Tolerance Factor (t) in Pb-I lattice | Key Optical Property | Thermal Degradation Pathway |
|---|---|---|---|---|
| Cesium (Cs⁺) | ~1.88 | ~0.89 [11] | Wider bandgap, stable blue emission [9] | Phase transition from γ-phase to δ-phase [9] |
| Formamidinium (FA⁺) | ~2.2-2.8 | ~0.99-1.06 [7] | Narrow bandgap, longer carrier lifetime [7] | Direct decomposition to PbI₂ [9] |
The Role and Stability of B-Site Metal Ions (Pb²⁺)
The B-site cation, typically lead (Pb²⁺), is the cornerstone of the perovskite octahedral structure. It forms strong covalent bonds with the surrounding halide anions, creating the [PbX₆]⁴⁻ octahedra that define the electronic band structure of the material. Pb²⁺ is responsible for the characteristic defect tolerance of lead halide perovskites, where certain intrinsic defects do not form deep-level traps within the bandgap [12] [11].
Compared to halide anions and A-site cations, the Pb²⁺ ion is significantly less mobile due to its higher charge and larger migration energy barrier [8]. Its primary role is structural integrity rather than long-range migration. However, the detachment of surface ligands can create unsaturated "dangling bonds" on Pb²⁺ atoms, which act as surface trap states, quenching luminescence and accelerating degradation [8] [11]. Furthermore, the potential release of toxic Pb²⁺ upon environmental degradation of PQDs remains a major concern for commercial applications, driving research into stable encapsulation strategies and less toxic lead-free alternatives [12] [13].
Experimental Protocols for Studying Ion Dynamics
In Situ Characterization of Ion Migration and Stability
Purpose: To directly observe the real-time structural and optical changes in PQDs induced by ion migration under external stress (e.g., heat). Materials:
- PQD Sample: Colloidally synthesized CsₓFA₁₋ₓPbI₃ PQDs with full compositional range (x = 0 to 1) [9].
- Instrumentation: In-situ X-ray Diffractometer (XRD) equipped with a heating stage; In-situ Photoluminescence (PL) spectroscopy setup with temperature control; Thermogravimetric Analyzer (TGA) [9].
- Environment Control: Argon gas flow to prevent oxidative degradation during heating [9].
Methodology:
- Sample Preparation: Deposit a thin, uniform film of PQDs onto a suitable substrate (e.g., Pt, silicon wafer).
- Temperature Ramp: Place the sample in the characterization instrument and program a controlled temperature ramp from room temperature (e.g., 30°C) to 500°C at a defined rate (e.g., 5-10°C/min).
- Simultaneous Data Collection:
- In-situ XRD: Continuously collect XRD patterns throughout the heating process. Monitor the appearance, disappearance, and shift of diffraction peaks corresponding to the perovskite black phase (γ/α), non-perovskite yellow phase (δ), and PbI₂ [9].
- In-situ PL: Track changes in the PL intensity, peak position, and full width at half maximum (FWHM) to correlate structural changes with optoelectronic properties [9].
- TGA: Measure the sample's weight loss to identify the decomposition temperature of organic components (e.g., FA⁺, ligands) [9].
- Data Analysis: Correlate the data from all techniques to establish composition-dependent degradation mechanisms. For instance, determine that Cs-rich PQDs degrade via a γ-to-δ phase transition, while FA-rich PQDs with higher ligand binding energy decompose directly into PbI₂ [9].
Post-Synthetic Cation Exchange for Mixed A-Site PQDs
Purpose: To synthesize mixed A-site PQDs (e.g., Cs₁₋ₘFAₘPbI₃) with precise control over composition and optical properties via a cation-exchange reaction. Materials:
- Parent PQDs: Purified colloidal suspension of single-cation PQDs (e.g., CsPbI₃ PQDs) [7].
- Cation Source: Solution of formamidinium oleate (FA-OA) or a colloidal suspension of a different single-cation PQD (e.g., FAPbI₃ PQDs) [7].
- Solvents: Non-polar solvents like hexane or octane.
- Ligands: Oleic acid (OA) and Oleylamine (OAm) to maintain colloidal stability during exchange [7].
- Environment: Inert atmosphere (e.g., N₂ glovebox) to prevent degradation.
Methodology:
- Preparation: Characterize the parent PQDs (CsPbI₃) to determine initial absorption, PL, and size.
- Reaction Setup: In an inert environment, add a controlled stoichiometric amount of the FA⁺ cation source (FA-OA solution or FAPbI₃ PQD suspension) to the parent PQD solution [7].
- Kinetic Control: Stir the mixture at a controlled temperature (e.g., room temperature or slightly elevated). The exchange kinetics are regulated by factors like solvent polarity, ligand concentration, and temperature [7].
- Monitoring: Periodically extract aliquots and characterize them using UV-Vis absorption and PL spectroscopy to track the shift in the bandgap and emission peak toward longer wavelengths (red-shift) as FA⁺ incorporates into the lattice [7].
- Termination and Purification: Once the desired optical properties are achieved, terminate the reaction by adding an antisolvent (e.g., methyl acetate) to precipitate the mixed-cation PQDs. Centrifuge and redisperse the purified PQDs in a non-polar solvent [7].
Visualization of Ion Dynamics and Experimental Workflows
Pathways of Ion Migration in Perovskite Quantum Dots
The following diagram illustrates the primary migration pathways and key dynamic processes of ions within a PQD lattice and at its surface.
Diagram 1: Pathways of ion migration and degradation in PQDs, highlighting internal halide and A-site cation mobility, as well as surface processes like ligand detachment that lead to ion release and structural instability.
Workflow for Investigating PQD Ion Dynamics
This flowchart outlines a comprehensive experimental methodology for synthesizing PQDs, inducing ion exchange, and characterizing the resulting stability and ion dynamics.
Diagram 2: Integrated experimental workflow for probing ion dynamics in PQDs, from synthesis and modification to in-situ/ ex-situ characterization and data modeling.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for PQD Synthesis and Ion Exchange Studies
| Reagent / Material | Typical Function | Technical Explanation & Rationale |
|---|---|---|
| Cesium Carbonate (Cs₂CO₃) | A-site precursor for Cs⁺ cation. | Reacts with fatty acids (e.g., oleic acid) to form Cs-oleate, the common Cs⁺ source in hot-injection synthesis of CsPbX₃ PQDs [7]. |
| Formamidinium Acetate (FAAc) | A-site precursor for FA⁺ cation. | Used in cation-exchange reactions; the FA⁺ ion replaces a portion of Cs⁺ in pre-synthesized CsPbX₃ PQDs to form mixed-cation systems (Cs₁₋ₘFAₘPbX₃) [7]. |
| Lead Bromide/Iodide (PbBr₂/PbI₂) | B-site and halide precursor. | Provides Pb²⁺ and halide ions (Br⁻, I⁻) for the perovskite framework. The purity and stoichiometry are critical for controlling crystal growth and defect density [11]. |
| Oleic Acid (OA) / Oleylamine (OAm) | Surface capping ligands. | Dynamic ligands that passivate surface defects, control nanocrystal growth, and provide colloidal stability. Their weak binding is a primary source of instability [8] [11]. |
| Methyl Acetate / Methyl Benzoate | Antisolvent for purification and rinsing. | Polar solvents that precipitate PQDs or rinse films to remove excess ligands without dissolving the perovskite core. They can hydrolyze to provide short-chain ligands (e.g., acetate) for X-site ligand exchange [14]. |
| 2-Aminoethanethiol (AET) | Post-synthesis ligand for stabilization. | A short-chain, bidentate ligand with a strong affinity for Pb²⁺. Used in post-treatment to replace OA/OAm, creating a dense passivation layer that enhances stability against water and UV light [8]. |
| Potassium Hydroxide (KOH) | Alkaline catalyst for ligand exchange. | Used to create an alkaline environment that facilitates the hydrolysis of ester antisolvents (e.g., methyl benzoate), making the substitution of long-chain insulating ligands with short conductive ones more efficient and rapid [14]. |
The dynamics of halide anions, A-site cations, and B-site metal ions are fundamental to the performance and stability of perovskite quantum dots. While halide mobility presents challenges for spectral stability, A-site cation exchange offers a powerful tool for compositional and bandgap tuning. The relative immobility of the B-site cation underpins structural integrity but necessitates careful surface passivation.
Future research must focus on decoupling ion migration from device operation. This will involve the development of advanced ligand systems that strongly bind to the PQD surface, suppressing both halide vacancy formation and A-site cation migration [8] [14]. Furthermore, engineering core-shell heterostructures or implementing crosslinking strategies could provide a physical barrier to ion diffusion. The insights from studying lead-based systems must also guide the design of lead-free alternatives (e.g., based on Bi³⁺ or Sn²⁺) that inherently possess higher ionic migration barriers [12] [13]. A multidisciplinary approach, integrating precise synthesis, advanced in-situ characterization, and theoretical modeling, is essential to fully understand and control ionic migration dynamics, thereby unlocking the full commercial potential of perovskite quantum dots.
Ionic migration within perovskite quantum dots (PQDs) is a double-edged sword. While it can induce deleterious degradation mechanisms in optoelectronic devices, its precise control also offers avenues for enhancing material properties and functionalities. The dynamics of these ion movements are not random; they are fundamentally governed by the nanoscale surface topography of the PQDs. This whitepaper delves into the critical, yet often overlooked, relationship between the surface energy landscape—sculpted by crystal curvature and exposed facets—and the guided pathways of ion migration. Framed within a broader thesis on ionic migration dynamics at PQD surfaces, this analysis synthesizes recent research to provide a technical guide for controlling ion transport through intelligent surface design. Understanding these principles is paramount for researchers and scientists aiming to develop next-generation PQD-based devices with unparalleled performance and operational stability.
The Surface Energy Landscape of PQDs
The surface of a perovskite quantum dot is a dynamic interface where its crystalline structure terminates, creating an environment rich in unsaturated bonds and, consequently, elevated surface energy. This energy landscape is not uniform; it is primarily determined by two interconnected geometric factors: local curvature and crystallographic facet expression.
Surface Curvature: At the nanoscale, regions with high positive curvature, such as sharp edges and vertices, exhibit a lower coordination number for surface atoms compared to flat planes. This atomic under-coordination makes these sites high-energy "hot spots." The inherent thermodynamic drive to minimize total system energy makes these sites particularly susceptible to ion adsorption, desorption, and migration. Conversely, flatter regions or negative curvatures represent lower-energy pathways or stable resting points for mobile ions. This energy gradient directly pulls ions from high-curvature to low-curvature regions.
Crystallographic Facets: A PQD is enclosed by a set of distinct crystal planes, or facets, each with a unique atomic arrangement and surface energy. For instance, in formamidinium lead iodide (FAPbI3), the (100) and (111) facets display markedly different stabilities. Research has shown that the (100) facet is significantly more susceptible to degradation from environmental factors like moisture compared to the (111) facet [15]. This implies that the activation energy for ion migration (e.g., the vacancy-mediated diffusion of iodide ions) is facet-dependent. A surface dominated by higher-energy facets will generally provide a lower-energy barrier for ionic motion, facilitating faster migration, while stable, low-energy facets can effectively pin ions in place.
The interplay between curvature and facet creates a complex energy map across the PQD surface. This map acts as a template, directing the preferential movement of ions such as iodide (I⁻), lead (Pb²⁺), and organic cations (e.g., FA⁺ or MA⁺) along specific trajectories, thereby dictating the material's evolution under operational stress.
Quantitative Data on Facet-Dependent Stability
The influence of specific crystallographic facets on PQD stability is not merely theoretical; it is quantitatively demonstrated through controlled experiments. The strategic stabilization of certain facets has led to direct improvements in key device performance metrics.
The table below summarizes empirical data on facet-dependent properties and their outcomes in solar cell devices:
Table 1: Quantitative Data on Facet-Dependent Stability in Perovskite Solar Cells
| Perovskite Material | Targeted Facet | Experimental Strategy | Key Outcome | Device Performance Impact |
|---|---|---|---|---|
| FAPbI3 [15] | (111) | Used cyclohexylamine additive to selectively promote (111) facet growth. | 95% retention of initial performance after ∼2000 hours at 30-40% relative humidity (unencapsulated). | Power Conversion Efficiency (PCE) of 24%. |
| FAPbI3 [15] | (100) | Control sample without facet-controlling additive. | Higher degradation rate when exposed to moisture. | Lower stability and performance compared to (111)-facet dominated samples. |
| Dion-Jacobson 2D Perovskite [15] | N/A (Phase-Pure) | Tri-solvent engineering to achieve pure-phase, oriented grains. | 95% retention for >3000 hours in air at 85°C with 60-90% relative humidity. | PCE of 17.27% for MA-free DJ 2D PSC. |
The data clearly indicates that surfaces dominated by the (111) facet in FAPbI3 exhibit superior stability against moisture-induced degradation compared to (100)-facet dominated surfaces [15]. This enhanced stability is directly linked to a reduced propensity for ion migration and structural decomposition at the surface. Furthermore, achieving phase-purity and fine control over the quantum well structure in low-dimensional perovskites, as in the Dion-Jacobson phase, represents an advanced form of facet and grain boundary engineering that profoundly suppresses ionic migration, leading to exceptional long-term stability under harsh environmental testing [15].
Experimental Protocols for Investigating Ion Movement
To systematically study the connection between surface topography and ion migration, researchers employ a suite of advanced characterization and testing protocols. The following workflow outlines a typical integrated experimental approach.
Diagram 1: Integrated experimental workflow for investigating ion movement in PQDs.
Detailed Methodologies
1. Sample Synthesis and Facet Engineering
- Objective: To synthesize PQDs with controlled dominance of specific crystallographic facets.
- Protocol:
- Precursor Preparation: Prepare lead iodide (PbI₂) and formamidinium iodide (FAI) in suitable polar solvents (e.g., DMF/DMSO).
- Ligand-Assisted Reprecipitation: Rapidly inject the perovskite precursor into a non-solvent (e.g., toluene) containing coordinating ligands (e.g., oleic acid and oleylamine) under vigorous stirring to induce nucleation and growth of PQDs.
- Facet Control: Introduce facet-directing agents (e.g., cyclohexylamine with a boiling point of 134°C) into the non-solvent. The additive selectively binds to certain crystal planes, inhibiting their growth and promoting the dominance of target facets like (111) [15].
- Purification: Centrifuge the resulting colloidal solution to remove unreacted precursors and aggregates. The PQD pellet is then redispersed in an anhydrous solvent for further use.
2. Structural and Surface Analysis
- Objective: To characterize the crystal structure, facet composition, and surface morphology of the synthesized PQDs.
- Protocol:
- Transmission Electron Microscopy (TEM): Acquire high-resolution TEM (HRTEM) images to directly observe the crystal lattice, measure interplanar spacings, and identify the dominant facets enclosing the PQDs.
- X-Ray Diffraction (XRD): Perform XRD analysis on PQD films. The relative intensity of diffraction peaks (e.g., (100) vs. (111)) provides quantitative information on the preferred crystal orientation and facet distribution.
3. Ion Migration Assessment
- Objective: To directly probe the dynamics and pathways of ion migration.
- Protocol:
- Thermally Stimulated Current (TSC) Measurement: Place a PQD film between two electrodes. Heat the device at a constant rate while applying a DC bias. The resulting current, caused by the release of trapped ions, is measured. The temperature peaks in the TSC spectrum correspond to different ionic species and their activation energies for migration.
- Scanning Kelvin Probe Microscopy (SKPM): Use an atomic force microscope (AFM) with a conductive probe to map the surface potential of a PQD film under bias and/or illumination. Redistribution of mobile ions changes the local work function, which is detected as a shift in contact potential difference, allowing for visualization of ion migration pathways.
4. Device Fabrication and Operational Stability Testing
- Objective: To correlate surface-induced ion migration with device-level performance and degradation.
- Protocol:
- Solar Cell Fabrication: Fabricate PSCs in either n-i-p or p-i-n architecture. Spin-coat the synthesized PQDs as the active layer onto a charge transport layer (e.g., SnO₂). Deposit subsequent layers and metal electrodes as per standard procedures [15].
- Maximum Power Point (MPP) Tracking: Operate the encapsulated or unencapsulated devices at their MPP under continuous simulated sunlight (e.g., 1 sun illumination at AM 1.5G). Monitor the PCE as a function of time (e.g., over 1000-2000 hours) to assess operational stability [15]. The decay rate is a direct indicator of the robustness of the PQD surface against ion-migration-induced degradation.
The Scientist's Toolkit: Research Reagent Solutions
The experimental pursuit of stable PQDs requires a carefully selected toolkit of reagents and materials. The following table catalogues essential items used in the featured research for controlling surface properties and mitigating ion migration.
Table 2: Essential Research Reagents for PQD Surface and Ion Migration Studies
| Reagent/Material | Function/Description | Application in Research |
|---|---|---|
| Cyclohexylamine [15] | A high-boiling-point amine additive that acts as a facet-directing agent. | Selectively promotes the growth of the stable (111) facet in FAPbI3 perovskites, thereby reducing surface energy and enhancing moisture resistance. |
| Uracil [15] | A biomolecule used as a multi-functional binder and passivant. | Strengthens grain boundaries and effectively passivates surface defects, improving the mechanical and ionic stability of perovskite films. |
| Oleic Acid & Oleylamine | Common surface-capping ligands used in colloidal nanocrystal synthesis. | Coordinate to surface atoms during PQD growth, controlling size and shape, and providing initial stabilization against aggregation and oxidation. |
| Guanabenz Acetate Salt [15] | A chemical agent used for ambient-air fabrication. | Prevents perovskite hydration by shielding the surface from water molecules, obviating both anion and cation vacancies that facilitate ion migration. |
| β-poly(1,1-difluoroethylene) (β-pV2F) [15] | A polymer with an ordered dipolar structure. | Used in strain-engineering to stabilize the perovskite black phase and control energy alignment at interfaces, reducing ionic mobility under thermal cycling. |
| Alkyl Ammonium Iodide Salts [16] | Ligands used for surface exchange and passivation. | Employed in ligand exchange strategies to create a dense, conductive PQD film with well-passivated surfaces, crucial for high-efficiency solar cells. |
The surface energy landscape of perovskite quantum dots, defined by the intricate interplay of curvature and crystallographic facets, serves as the fundamental master planner for ionic migration. This whitepaper has established that a profound understanding of this relationship is not merely academic but is instrumental in developing actionable strategies for enhancing PQD stability. By deliberately engineering surfaces towards low-energy, stable facets like (111) in FAPbI3, and by employing sophisticated passivation and ligand engineering techniques, researchers can effectively construct energy barriers that suppress detrimental ion movement. The experimental frameworks and reagent tools outlined herein provide a pathway for continued exploration and optimization. As the broader thesis of ionic migration dynamics evolves, the deliberate design of the surface energy landscape will undoubtedly remain a central tenet, guiding the development of robust, high-performance PQD technologies that can finally transition from laboratory marvels to commercial realities.
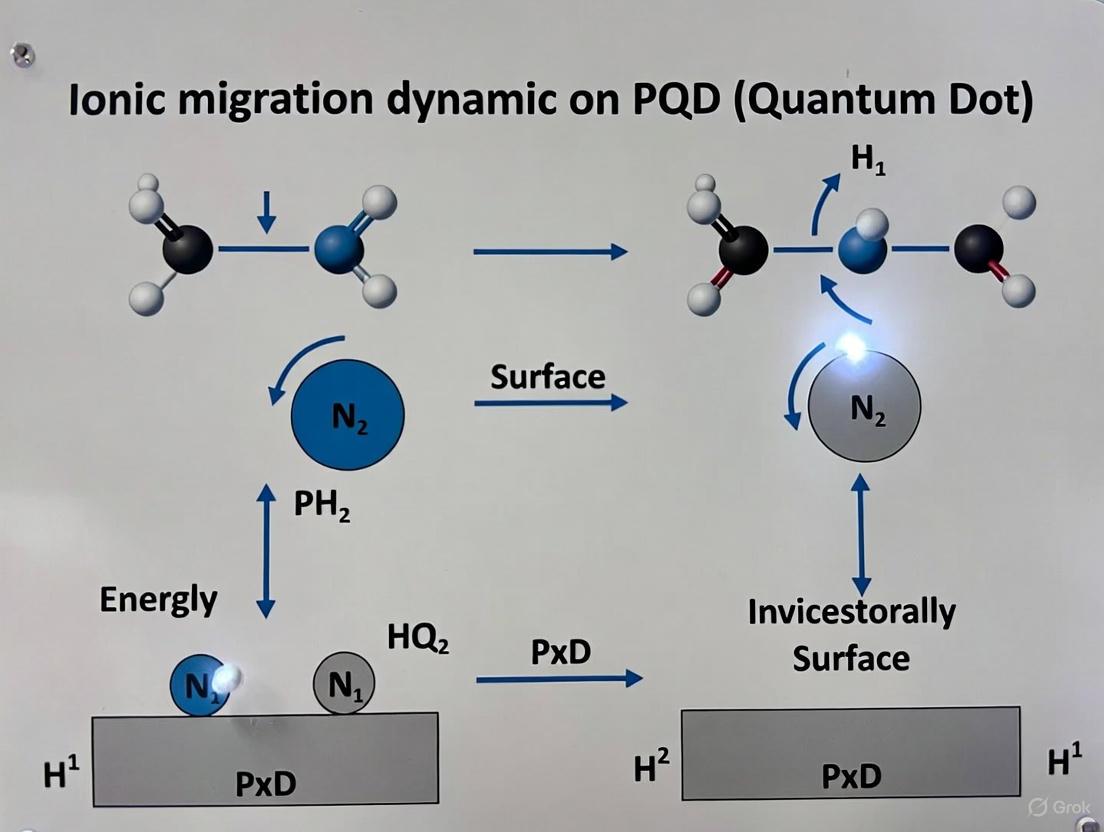
The operational stability and performance of perovskite quantum dots (PQDs) in optoelectronic devices are intrinsically limited by the complex interplay between ionic migration and surface defects. This technical guide delves into the atomistic origins of surface traps and their dynamic relationship with ion migration, a critical challenge identified in current research on PQD surfaces. While PQDs exhibit exceptional optoelectronic properties such as high photoluminescence quantum yields (PLQY) and tunable bandgaps, their intrinsic instability remains a major obstacle to commercialization [17]. This review synthesizes recent findings on the mechanisms of defect formation and ion migration, provides detailed methodologies for their experimental and computational investigation, and offers a toolkit for researchers aiming to mitigate these detrimental phenomena. The insights framed within this document contribute to the broader thesis that understanding and controlling these interfacial dynamics is paramount for developing next-generation, stable perovskite-based technologies.
Low-dimensional halide perovskites, including quantum dots, nanowires, and nanosheets, hold significant promise for optoelectronic applications due to their distinctive quantum confinement effects, adjustable bandgaps, and superior carrier dynamics [17]. However, their practical application is severely constrained by two interrelated phenomena: surface defects and ionic migration. Surface traps, which are ubiquitous in nanoscopic semiconductor materials, originate from the lower coordination of surface atoms compared to bulk atoms, leading to localized electronic states that act as centers for non-radiative recombination [18]. Concurrently, the soft, ionic lattice of perovskite materials facilitates the migration of ions (such as halide anions and A-site cations) under operational stresses like electric fields and light illumination.
The interplay between these processes creates a feedback loop: ionic migration can modify the surface chemistry and passivation of PQDs, leading to the formation of new trap states, while existing surface defects can act as preferential sites for ion accumulation and nucleation of degradation. This synergy ultimately accelerates the degradation of perovskite optoelectronic devices, impacting their efficiency and operational lifetime [17]. Understanding this coupling is therefore not merely an academic exercise but a critical requirement for engineering robust materials.
Fundamental Mechanisms and Atomistic Origins
Classification and Chemistry of Surface Defects
Surface traps can be classified as either shallow or deep midgap states, with the latter being particularly detrimental for device performance as they provide efficient pathways for non-radiative exciton recombination [18]. The atomistic origin of these traps is best understood by considering specific benchmark systems:
- In CsPbI₃ PQDs: The surface chemistry is highly dynamic. Trap states can form due to the displacement of surface ions or the adsorption/desorption of ligand species. Computational models using charge-balanced chemical formulas like [ABX₃]ₘ(AX)ₙ are crucial for reliably simulating these systems without introducing spurious electronic states from unrealistic stoichiometries [18].
- In PbS QDs: The formation of trap states is often linked to non-stoichiometric surfaces. For instance, the removal of two iodide ligands to form molecular iodine can lead to an n-doped QD (Ndop = -2 in the Charge Balance Model), with excess electrons filling the conduction band and creating a doped system that differs from the intrinsic, defect-free material [18].
The Charge Balance Model (CBM) and Covalent Bond Classification (CBC) scheme provide complementary frameworks for describing the QD's electronic structure and surface chemistry, respectively [18]. The CBM evaluates charge balance by treating the QD as an ionic species, counting the number of excess or deficient valence electrons. A condition of Ndop = 0 typically indicates an intrinsic, charge-balanced QD with a clean band gap. The CBC scheme, conversely, describes the chemical bonding at the surface using neutral species and classifies ligands as L-type (2-electron donors), X-type (1-electron donors), or Z-type (0-electron donors, often Lewis acidic metal complexes) [18]. Proper ligand passivation, often using a combination of these types, is essential for achieving a stable, trap-free surface.
The Ionic Migration Pathway
Ionic mobility (μ) or diffusivity (D) in a crystalline solid is exponentially dependent on the migration barrier (Em) for ionic motion, as described by the equation: D = f·g·a²·ν·exp(-Em/kBT) [19]
Here, f is the correlation factor, g is the geometric factor describing diffusion channel connectivity, a is the hop distance, ν is the pre-factor dependent on vibrational frequencies, and kB and T are the Boltzmann constant and temperature, respectively. Among these factors, the migration barrier (Em) is the most dominant, as it has an exponential influence on diffusivity [19]. Identifying materials with high ionic mobility therefore hinges on accurately predicting and minimizing Em.
Table 1: Key Factors Influencing Ionic Migration Barrier (Em) in Solids
| Factor | Description | Impact on Migration Barrier (Em) |
|---|---|---|
| Crystal Structure & Connectivity | Geometric factor (g) and hop distance (a); connectivity of diffusion channels. | Em is often lower in structures with open, connected pathways and optimal transition state geometry that avoids face-sharing polyhedra [19]. |
| Migrating Ion-Anion Distance | The distance between the migrating ion and the surrounding anions in the lattice. | Shorter distances often lead to higher Em due to stronger repulsive interactions during the hop [19]. |
| Coordination Environment Change | The change in coordination number of the migrating ion between its initial and transition states. | A significant change in coordination number typically leads to a higher Em [19]. |
Ionic migration in perovskites is not a random walk but occurs through specific pathways within the crystal lattice, often involving vacancy-mediated mechanisms. The energy landscape for these pathways is dictated by the surrounding lattice and the local chemical environment, which can be significantly altered by the presence of surface defects.
Coupling Dynamics: How Defects and Migration Interact
The relationship between vacancies, traps, and ion migration is cyclic. First, vacancies are a prerequisite for ion migration; without vacant lattice sites, ions cannot readily hop. Second, the migration of ions can itself generate defects. For example, when an ion migrates from a lattice site to an interstitial position, it creates a vacancy-interstitial pair (Frenkel defect). If the migrating ion is subsequently trapped at the surface, it can leave behind a vacancy cluster in the bulk. Third, existing surface defects act as sinks for migrating ions. Ions such as I⁻ can accumulate at surface trap sites, leading to localized chemical decomposition, such as the formation of PbI₂, which is a common degradation product observed in lead-halide perovskites [17]. This accumulation not only passivates the trap state but also creates new ones, altering the surface's electronic structure and potentially triggering further ion migration to restore local charge balance.
Experimental and Computational Characterization Methods
A multi-pronged approach is required to probe the complex interplay between ionic migration and surface defects. The following workflows and protocols outline key methodologies.
Computational Workflow for Predicting Migration Barriers
Diagram 1: Workflow for computing ionic migration barriers.
Detailed Protocol: Density Functional Theory - Nudged Elastic Band (DFT-NEB) Calculation
The DFT-NEB method is considered the state-of-the-art for accurately estimating Em by modeling the minimum energy path (MEP) of atomic migration [19].
- Supercell Construction: Build a crystallographic supercell of the material of sufficient size to prevent interaction between periodic images of the defect/vacancy. A 2x2x2 or 3x3x3 supercell is typical.
- Geometry Optimization: Use DFT to fully relax the atomic positions and lattice vectors of the supercell to its ground-state configuration. This provides the initial (IS) and final (FS) states for the migration hop.
- Pathway Initialization: Construct a series of intermediate "images" (typically 5-9) between the IS and FS. A simple linear interpolation of atomic coordinates is often used for initial path guess.
- NEB Calculation: Perform the NEB calculation using a DFT code (e.g., VASP, Quantum ESPRESSO). The images are connected by spring forces and optimized simultaneously. The climbing-image NEB variant is recommended to ensure the highest-energy image converges to the saddle point.
- Energy Barrier Extraction: The migration barrier (Em) is calculated as the total energy difference between the saddle point image and the initial state image.
Protocol Notes: This method is computationally intensive, with cost scaling with system size. The choice of exchange-correlation functional can impact accuracy. For high-throughput screening, machine learning models like graph neural networks (GNNs) fine-tuned on DFT-NEB data can predict Em swiftly and accurately, achieving R² scores >0.7 on diverse test sets [19].
Experimental Workflow for Trap State Analysis
Diagram 2: Experimental characterization of traps and ion migration.
Detailed Protocol: Photoluminescence Quantum Yield (PLQY) and Lifetime Decay
PLQY is a direct indicator of the efficiency of radiative recombination and, by extension, the density of non-radiative trap states [18].
- Sample Preparation: Prepare a stable, optically clear dispersion of PQDs in a suitable solvent. Ensure the optical density at the excitation wavelength is low (e.g., <0.1) to avoid inner-filter effects.
- Absolute PLQY Measurement: Use an integrating sphere coupled to a spectrometer and a calibrated light source. The sample is excited inside the sphere.
- Measure the spectrum of the excitation light with only the solvent in place (Iex(λ)).
- Measure the spectrum with the PQD sample in place. This contains both the transmitted excitation light and the photoluminescence (Iem(λ)).
- The absolute PLQY is calculated as: PLQY = ∫Iem(λ)dλ / [∫Iex(λ)dλ - ∫Isample(λ)dλ], where Isample(λ) is the transmitted excitation light measured with the sample.
- Time-Resolved PL (TRPL) Measurement: Use a time-correlated single photon counting (TCSPC) system with a pulsed laser source (e.g., picosecond diode laser).
- Record the decay of photoluminescence intensity after pulsed excitation.
- Fit the decay curve to a multi-exponential model: I(t) = Σ Aᵢ exp(-t/τᵢ). The amplitude-weighted average lifetime (⟨τ⟩ = Σ Aᵢτᵢ / Σ Aᵢ) is often reported.
- Interpretation: A high PLQY (approaching 100%) and a long average PL lifetime are indicative of low trap state density and effective surface passivation [17]. A multi-exponential decay suggests a distribution of carrier dynamics, often due to trapping and detrapping from surface states.
Protocol Notes: For measuring ionic mobility, Electrochemical Impedance Spectroscopy (EIS) is a key technique, though it can be resource-intensive and sensitive to sample preparation [19].
Table 2: Key Characterization Techniques for Defects and Ion Migration
| Technique | Measured Property | Information on Defects/Migration | Key Considerations |
|---|---|---|---|
| Time-Resolved PL (TRPL) | Photoluminescence decay lifetime | Dynamics of charge carrier trapping and non-radiative recombination; trap-assisted recombination rates. | Requires multi-exponential fitting; sensitive to surface chemistry. |
| FTPS/SPS (Frequency/Photo-thermal Deflection Spectroscopy) | Sub-bandgap absorption | Density and energy distribution of trap states within the bandgap. | Highly sensitive to shallow and deep traps; requires specialized setup. |
| Electrochemical Impedance Spectroscopy (EIS) | Complex impedance as a function of frequency | Ionic conductivity, diffusion coefficients, and activation energy for migration (Em). | Analysis can be complex; requires modeling with equivalent circuits. |
| DFT-NEB Calculations | Total energy along a migration path | Atomistic migration pathway and energy barrier (Em). | Computationally expensive; accuracy depends on exchange-correlation functional. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for PQD Surface Defect and Ion Migration Research
| Reagent / Material | Function / Role | Example in Context |
|---|---|---|
| Lead Precursors (e.g., Pb(OAc)₂, PbI₂) | Source of Pb²⁺ cations for the perovskite inorganic framework. | PbI₂ is a common precursor for CsPbI₃ QD synthesis via hot injection [17]. |
| Cesium Oleate | Source of Cs⁺ cations. | Injected into lead-halide precursor solution to nucleate and grow CsPbX₃ QDs [17]. |
| Oleic Acid & Oleylamine | L-type and X-type ligands. | Primary surface ligands that passivate under-coordinated surface atoms (Pb²⁺ and I⁻) during synthesis, suppressing trap formation [18]. |
| Halide Salt Precursors (e.g., NH₄X, ZnX₂) | Source of halide anions (X = Cl, Br, I); used for anion exchange. | Enables post-synthetic tuning of bandgap and PL emission [17]. Can also be used to treat halide-deficient surfaces. |
| Bidentate Ligands (e.g., 2-Bromohexadecanoic Acid - BHA) | Advanced surface passivators. | Bidentate ligands bind more strongly to surface atoms, effectively passivating surface defects and leading to high PLQY (>97%) even under prolonged UV irradiation [17]. |
| Z-type Passivators (e.g., CdX₂, PbX₂) | Electron-withdrawing Lewis acid species. | Passivate under-coordinated halide anions (Lewis bases) on the QD surface, as described by the CBC scheme, helping to achieve charge balance [18]. |
| Metal Halide Salts (e.g., KI, ZnBr₂) | Post-synthetic healing agents. | Used in solution or solid-state treatments to fill halide vacancies, a common defect that acts as a trap and facilitates I⁻ migration [18]. |
The intricate dance between ionic migration and surface defects lies at the heart of the stability challenge in perovskite quantum dots. As detailed in this guide, surface traps originating from under-coordinated atoms and non-stoichiometric surfaces create midgap states that quench luminescence and degrade performance. Simultaneously, the low migration barriers for ions in the soft perovskite lattice enable a dynamic redistribution of species that can both create new defects and be guided by existing ones. Breaking this detrimental cycle requires a holistic approach that combines advanced synthesis with precise passivation and thoughtful material design.
Future research must focus on several key areas:
- Developing Unified Models: Creating computational models that can simultaneously describe electronic structure (defects) and ion dynamics, bridging the gap between static and dynamic descriptions of the PQD surface.
- In Situ/Operando Characterization: Applying techniques like in situ spectroscopy and diffraction under operational conditions (light, bias) to observe the interplay between defects and migration in real-time.
- Multi-modal Passivation Strategies: Designing and implementing robust passivation schemes that use a combination of L-, X-, and Z-type ligands to comprehensively address all possible surface defect types, thereby stabilizing the surface against both ionic attack and ligand loss [18].
- Exploiting Machine Learning: Leveraging accurate graph neural network models and other ML tools to rapidly screen for new compositions and heterostructures with inherently low ion migration barriers and high defect tolerance, accelerating the discovery of stable materials [19].
By deepening our understanding of the fundamental mechanisms outlined in this review and leveraging the detailed protocols and tools provided, researchers can make significant strides toward overcoming the primary barriers to the commercialization of high-performance, durable perovskite quantum dot technologies.
Ionic migration, the movement of charged atoms or molecules, is a fundamental process governing functionality in a wide range of advanced materials, from energy storage systems to novel sensing platforms. Within the specific context of perovskite quantum dot (PQD) surfaces research, controlling this migration is paramount for enhancing device performance and stability. External stimuli such as light, electric fields, and heat provide powerful, non-invasive means to precisely activate and direct ion transport. This control enables the tuning of material properties on-demand, facilitating breakthroughs in applications like photovoltaics, chemical sensing, and bioelectronic medicine. A deep understanding of these triggers is not merely an academic exercise but a practical necessity for designing the next generation of adaptive and high-performance technologies. This whitepaper provides an in-depth technical examination of the mechanisms and methodologies by which these external triggers govern ionic movement, framed within the ongoing research on ionic migration dynamics at PQD surfaces.
The Role of Light in Ion Transport
Light energy, particularly in the visible spectrum, can induce ion transport through direct photon-matter interactions that alter the energy landscape within a material. A prominent mechanism involves light-induced ligand dissociation in metal-organic complexes.
Mechanism of Visible-Light-ContMetal-Ligand Coordination
Research has demonstrated that Ru(II) complexes, such as [Ru(tpy-COOH)(biq)(H2O)](PF6)2 (denoted Ru-H2O), can undergo reversible ligand substitution controlled by visible light [20]. In this system:
- Thermal Substitution (in the dark): A coordinated water molecule in the Ru-H2O complex can be replaced by a functional thioether ligand (e.g., MeSC2H4-R1), forming a stable Ru–thioether coordination bond.
- Photosubstitution (under light): Upon irradiation with visible light (e.g., 530 nm at 50 mW cm⁻²), the coordinated thioether ligand is cleaved and replaced by a water molecule [20].
This reversible process, cyclable over at least 10 times with approximately 80% coordination efficiency per cycle, effectively acts as a molecular "screwdriver" for reconfiguring surface functions and controlling ionic species [20]. The equilibrium constant (K) for the coordination reaction with a model thioether like 2-(methylthio)ethanol (MTE) is 107 ± 4 M⁻¹ at 298 K, indicating a strong, yet reversible, binding affinity [20].
Experimental Protocol: Light-Induced Ligand Exchange on Surfaces
Objective: To functionalize and subsequently reconfigure a surface using visible-light-controlled Ru–thioether coordination.
Materials:
- Quartz or silicon substrate.
- (3-aminopropyl)triethoxysilane (APTES).
- Ru-H2O complex.
- Coupling agents: N-Hydroxysuccinimide (NHS) and N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC).
- Aqueous solutions of functional thioether ligands (e.g., MeSC2H4-R1, MeSC2H4-R2).
- Solvents: Water, acetone.
Procedure:
- Substrate Preparation: Clean the substrate thoroughly (e.g., with oxygen plasma or piranha solution) to ensure a hydrophilic surface.
- Silanization: Immerse the substrate in a 2% v/v solution of APTES in toluene to form an amine-terminated surface. Rinse and cure.
- Ru Complex Grafting: React the aminated surface with the Ru-H2O complex using standard NHS/EDC carbodiimide chemistry to form amide bonds, creating a
Ru-H2O-modified substrate[20]. - Initial Functionalization (Thermal Substitution): Immerse the Ru-H2O-modified substrate in an aqueous solution of the first functional thioether (MeSC2H4-R1, 10 mM) for 40-60 minutes in the dark. Rinse gently with water to remove unbound ligand.
- Ligand Removal (Photosubstitution): Irradiate the functionalized surface with green light (530 nm, 40-50 mW cm⁻²) for 10 minutes in an aqueous environment to cleave the thioether ligand. Wash with water and acetone to remove the dissociated ligand.
- Re-functionalization (Thermal Substitution): Immerse the substrate in a solution of a second functional thioether (MeSC2H4-R2, 10 mM) in the dark for 40-60 minutes to attach the new functionality. Rinse.
Characterization: The success of each functionalization and de-functionalization step can be monitored using UV-vis absorption spectroscopy, observing the characteristic blueshift in the metal-to-ligand charge transfer band upon thioether coordination [20].
The following diagram illustrates the experimental workflow for reconfiguring a surface using this light-controlled coordination chemistry.
The Role of Electric Fields in Ion Transport
Electric fields provide a direct means to exert force on charged particles, enabling precise spatial and temporal control over ion transport. This principle is leveraged in both sensing and bioelectronic applications.
Mechanism of Ion-Selective Membrane Cuff Operation
A key innovation is the multimodal ion-selective membrane (ISM) cuff for focused ion depletion in vivo. The operating principle involves:
- Ion Depletion: When a current is applied across an ISM that is selective for a specific ion (e.g., Ca²⁺), the target ion is depleted in the electrolyte volume where current enters the membrane [21].
- Confinement: The cuff architecture physically confines the electrolyte volume around a nerve, focusing the ion depletion effect and preventing rapid dissipation into surrounding tissue. A transport model predicted that applying -20 µA for 5 minutes could reduce Ca²⁺ concentration in the cuff's lumen from 2 mM to below 0.5 mM [21].
- Nerve Sensitization: Depleting extracellular Ca²⁺ concentrations affects the gating of voltage-gated sodium channels, sensitizing the nerve and lowering its threshold for activation by electrical stimulation [21].
Experimental Protocol: In Vivo Nerve Sensitization via Focused Ca²⁺ Depletion
Objective: To modulate the sensitivity of a sciatic nerve to electrical stimulation in a live rat model using an ISM-cuff for localized Ca²⁺ depletion.
Materials:
- Implantable ISM cuff electrode (screen-printed carbon-polymer contacts, one coated with a Ca²⁺-selective membrane).
- Surgical setup for rat sciatic nerve exposure.
- Bipolar electrode for electrical stimulation.
- Recording equipment for compound muscle action potentials (CMAPs) from target muscles (e.g., gastrocnemius, tibialis anterior).
Procedure:
- Device Fabrication: Fabricate the cuff electrode with carbon-polymer contacts. Coat one contact with a Ca²⁺-selective membrane cocktail containing an ionophore [21].
- Surgical Implantation: Anesthetize the rat and acutely expose the sciatic nerve in the gluteal region. Carefully place the ISM cuff around the nerve.
- Baseline Threshold Search: Prior to ion modulation, determine the baseline threshold current required to elicit a CMAP in the target muscles using the cuff's uncoated stimulation electrode.
- Ion Depletion Phase: Apply a depletion current of -20 µA to the ISM-coated contact for 5 minutes. The transport model and direct measurements confirm this reduces Ca²⁺ to ~0.5 mM within the cuff lumen [21].
- Post-Depletion Threshold Search: Immediately after the depletion phase, repeat the threshold search from Step 3 to measure the change in nerve sensitivity.
- Data Analysis: Compare the stimulation thresholds and CMAP amplitudes before and after Ca²⁺ depletion to quantify the sensitization effect. Evidence suggests this effect can selectively influence different nerve fascicles, improving functional selectivity [21].
Characterization: The Ca²⁺ depletion performance of the ISM cuff can be directly measured in vitro using a calibrated sensing technique, verifying a drop from 2 mM to 0.5 mM within 100 s of applying -20 µA [21].
The diagram below outlines the key components and operating principle of the ISM-cuff device.
The Role of Heat in Ion Transport
Thermal energy influences ion transport by increasing ionic mobility and, in some material systems, inducing phase transitions that create new transport pathways or alter existing ones.
Mechanism of Thermal Phase Evolution in Hollandite Nanorods
In situ transmission electron microscopy (TEM) studies on silver hollandite (AgyMn8O16) nanorods have visualized the phase evolution and ion transport dynamics during lithiation, processes highly influenced by thermal conditions.
- Two-Stage Lithiation: The process involves two distinct regimes:
- β-regime: Characterized by fast Li⁺ diffusion along the nanorod's long axis with minimal volume change, resulting in a lithiated phase with an orthorhombic distortion (
LixAg1.6Mn8O16, x~1) [22]. - γ-regime: Initiated by a slower-moving reaction front, this stage involves substantial volume expansion (over 27% radially), the formation of polyphase lithiated hollandite, and the expulsion of face-centered-cubic silver metal (Ag⁰) nanoparticles (x>6 in
LixAg1.6Mn8O16) [22].
- β-regime: Characterized by fast Li⁺ diffusion along the nanorod's long axis with minimal volume change, resulting in a lithiated phase with an orthorhombic distortion (
- Inter-Nanorod Transport: A key finding was the first direct observation of lateral Li⁺ transport between individual nanorods in the a–b plane, not just along a single rod's c-axis. This indicates that ion transport in one-dimensional materials is not necessarily confined to a single particle [22].
Experimental Protocol: In Situ TEM Visualization of Li⁺ Transport
Objective: To observe in real-time the lithium-ion transport pathways and associated phase evolution within and between Ag1.6Mn8O16 nanorods.
Materials:
- Synthesized
Ag1.6Mn8O16nanorods (e.g., via hydrothermal or reflux-based synthesis). - In situ TEM scanning/transmission electron microscope (S/TEM) holder equipped with a nanomanipulator and a lithium metal tip.
- Lithium metal counter electrode.
- Synthesized
Procedure:
- Sample Preparation: Disperse the synthesized
Ag1.6Mn8O16nanorods in a solvent and drop-cast them onto a specialized in situ TEM chip with micro-electrodes. - Setup Assembly: Load the chip into the TEM holder. Using the nanomanipulator, bring a sharp lithium metal tip into contact with a selected nanorod or a bundle of nanorods.
- In Situ Lithiation: Apply a small bias potential between the Li tip (anode) and the chip's electrode (cathode) to initiate electrochemical lithiation while the holder is inside the TEM column.
- Real-Time Imaging & Diffraction: Record real-time video (e.g., 1 frame per second) to monitor morphological changes, such as the propagation of reaction fronts and volume expansion. Simultaneously, acquire selected area electron diffraction (SAED) patterns and electron energy-loss spectroscopy (EELS) spectra from specific regions (pristine, β-regime, γ-regime, reaction front) to correlate structural and chemical evolution [22].
- Data Analysis: Measure the velocity of reaction front propagation (observed at ~5 nm/s along the c-axis). Quantify volume changes from image analysis. Identify crystalline phases present in different regimes from diffraction patterns.
- Sample Preparation: Disperse the synthesized
Characterization: Key metrics include reaction front velocity, radial expansion percentage, and identification of crystalline phases (tetragonal hollandite, distorted lithiated phases, fcc Ag⁰) via EDPs [22].
The following tables consolidate key quantitative findings from the research discussed in this whitepaper.
Table 1: Performance Metrics of External Trigger Mechanisms
| Trigger Mechanism | Key Metric | Value / Range | Experimental Context |
|---|---|---|---|
| Light (Ru-Complex) | Photosubstitution Efficiency | ~80% per cycle | Reversible coordination of MTE to Ru-H2O over 10 cycles [20] |
| Equilibrium Constant (K) | 107 ± 4 M⁻¹ | Coordination of MTE with Ru-H2O at 298 K [20] | |
| Irradiation Parameters | 530 nm, 50 mW cm⁻², 1 min (soln) / 10 min (surface) | Green light-induced ligand dissociation [20] | |
| Electric Field (ISM Cuff) | Depletion Current | -20 µA | Current applied to ISM contact for Ca²⁺ depletion [21] |
| Ca²⁺ Concentration Reduction | 2.0 mM → <0.5 mM | In the cuff lumen after 5 min of -20 µA current [21] | |
| Electrode Impedance | 341 ± 36.9 Ω | Screen-printed carbon-polymer contacts [21] | |
| Heat (Ion Transport) | Reaction Front Velocity | ~5 nm/s | Propagation along nanorod c-axis during lithiation [22] |
| Volume Expansion (Radial) | >27% | In γ-regime of lithiated hollandite nanorods [22] | |
| Lithiation Stoichiometry (x in LixAg1.6Mn8O16) | β-regime: x~1; γ-regime: x>6 | Determined via EELS analysis [22] |
Table 2: Research Reagent Solutions for Featured Experiments
| Reagent / Material | Function / Role | Experimental Context |
|---|---|---|
[Ru(tpy-COOH)(biq)(H2O)](PF6)2 (Ru-H2O) |
Photoswitchable molecular "screwdriver"; core complex for reversible ligand coordination. | Light-controlled surface reconfiguration [20] |
| Functional Thioethers (MeSC2H4-R) | Molecular "bits"; provide specific surface functionalities (e.g., wettability, protein affinity). | Light-controlled surface reconfiguration [20] |
| Ca²⁺-Selective Ionophore Membrane | Selectively filters and depletes Ca²⁺ ions from a mixed electrolyte upon application of current. | Electrochemical nerve sensitization [21] |
| Screen-Printed Carbon-Polymer Contacts | Biocompatible, low-impedance electrodes for electrical stimulation and iontronic delivery in vivo. | ISM Cuff fabrication [21] |
| Silver Hollandite (Ag1.6Mn8O16) Nanorods | Tunnel-structured electroactive material for visualizing Li⁺ transport and phase evolution. | In situ TEM lithiation studies [22] |
Integrated Trigger Effects and Research Applications
The interplay of multiple external triggers often yields synergistic effects. For instance, the fabrication of perovskite quantum dots (PQDs) themselves, which are highly relevant to ionic migration surface research, often involves heat (e.g., hot-injection synthesis at elevated temperatures) to control crystallization and defect formation [12]. These PQDs, particularly lead-based variants like CsPbX3, are then deployed as nanosensors, where their interaction with light (high photoluminescence quantum yield) is modulated by the presence of target ions, enabling ultrasensitive heavy metal detection with limits as low as 0.1 nM [12]. Furthermore, the performance of related devices, such as perovskite solar cells (PSCs), is enhanced by strategies that manage ion migration under operational stresses combining electric fields and heat, achieving remarkable stabilities exceeding 8 months and power conversion efficiencies over 26% [15].
The fundamental insights and advanced methodologies detailed in this whitepaper—from light-controlled molecular reconfiguration to in situ visualization of ion transport—provide a powerful toolkit for researchers. These approaches are directly applicable to the broader thesis of understanding and controlling ionic migration dynamics at the surfaces of perovskite quantum dots and other advanced functional materials, paving the way for innovations in sensing, energy storage, and bioelectronic medicine.
Tools and Techniques: Probing and Harnessing Surface Ion Dynamics
Advanced Spectroscopy for Direct Ionic Motion Observation
Ionic motion, the migration of ions within a material's lattice or across its surface, is a fundamental process that dictates the performance and stability of numerous advanced technologies. In the context of perovskite quantum dots (PQDs)—a class of materials with exceptional optoelectronic properties—controlling ionic migration is a central challenge for achieving commercial viability. The inherent ionic nature of perovskite crystals, characterized by relatively low ionic migration energy barriers, facilitates the easy formation of halide vacancies and leads to structural degradation under external stimuli such as heat, light, or electrical bias [23]. This ionic mobility, while potentially exploitable in certain applications, often results in phase segregation, accelerated aging, and diminished device performance in solar cells and light-emitting diodes (LEDs) [14] [23]. Directly observing these ionic dynamics is therefore not merely an academic exercise but a critical endeavor for diagnosing failure mechanisms and guiding the rational design of more robust materials.
This technical guide details advanced spectroscopy techniques capable of probing ionic motion directly, with a specific focus on methodologies relevant to PQD surfaces. The ability to track ion movement with high spatial and temporal resolution provides invaluable insights into the fundamental dynamics that underpin device operation and degradation. For PQDs, surface ions are particularly susceptible to migration due to incomplete passivation and the presence of dangling bonds, making the surface a primary site for initiating structural failure [23]. The techniques outlined herein enable researchers to move beyond indirect performance metrics and observe the core ionic processes in real-time, thereby framing the discussion within the broader thesis of understanding and controlling ionic migration dynamics in PQD research.
Core Spectroscopy Techniques
A range of sophisticated spectroscopic methods has been developed to characterize ionic motion, each with unique mechanisms, advantages, and operational domains. The following table summarizes the key techniques of relevance to PQD studies.
Table 1: Core Spectroscopy Techniques for Observing Ionic Motion
| Technique | Fundamental Principle | Key Measurable | Spatial/Temporal Resolution | Primary Application in PQD Research |
|---|---|---|---|---|
| Ion Mobility Spectrometry-Mass Spectrometry (IMS-MS) | Separates ions in a buffer gas under an electric field based on their size, charge, and shape [24]. | Collision Cross Section (CCS, Ω), a measure of ionic gas-phase size [24]. | Millisecond separation timescale; compatible with chromatographic pre-separation [24]. | Probing the surface ligand shell and identifying labile ionic species desorbed from PQD surfaces [24]. |
| Impedance Spectroscopy (IS) | Applies a small AC voltage and measures the current response across a frequency range to deconvolute different electrochemical processes [25]. | Ionic conductivity, defect relaxation times, and interfacial charge transfer resistances [25]. | Bulk measurement; frequency-dependent, typically mHz to MHz range [25]. | Quantifying bulk ionic conductivity and characterizing the energy of ion migration within PQD films [25]. |
| Precision Laser Spectroscopy | Uses quantum logic with co-trapped atomic ions to prepare, control, and measure the quantum states of a single molecular ion [26]. | Rovibrational state transitions, dipole moments, and state lifetimes with ultra-high precision [26]. | Quantum-state resolution; sub-part-per-trillion spectral resolution achievable [26]. | Fundamental studies of ion-molecule interactions and electric field effects on single ions, providing benchmark data [26]. |
Technical Deep Dive: IMS-MS Methodologies
Among these techniques, IMS-MS deserves particular attention for its ability to separate and identify ionic species directly. The core principle, as defined by the Mason-Schamp equation, links the measured ion mobility (K) to the collision cross section (CCS, Ω), which is a normalized measure of the ion's gas-phase size [24]. The equation is:
[ \Omega = \frac{3}{16} \frac{ze}{N0} \left( \frac{2\pi}{\mu kB T} \right)^{1/2} \frac{1}{K_0} ]
Here, (ze) is the ion charge, (N0) is the buffer gas density, (\mu) is the reduced mass of the ion and buffer gas, (kB) is Boltzmann's constant, and (T) is the drift region temperature [24]. This allows researchers to distinguish between isomeric species and conformational states that would be indistinguishable by mass alone, making it powerful for analyzing the complex mixture of ligands and surface-bound ions on PQDs.
Different IMS platforms offer distinct advantages:
- Drift-Tube IMS (DTIMS): Considered the "classic" model, DTIMS uses a uniform electric field in a pressurized drift region. Its primary advantage is the ability to measure CCS as a primary method from first principles using the Mason-Schamp equation, without requiring calibration [24]. However, its pulsed nature can lead to a low duty cycle (e.g., ~6.7%), though multiplexing strategies like the Hadamard Transformation can increase this to ~50% [24].
- Traveling Wave IMS (TWIMS): This platform, which popularized IMS-MS, uses a series of dynamic voltage waves to propel ions through the gas. While it offers continuous ion introduction and high sensitivity, it is a secondary method that requires calibration with ions of known CCS values measured by DTIMS [24].
Experimental Protocols for Key Techniques
Protocol: Impedance Spectroscopy for Ionic Conductivity
Impedance Spectroscopy is a cornerstone technique for quantifying ionic motion in solid-state materials like perovskite films. A harmonized procedure is critical for obtaining reproducible and reliable results, as highlighted in a cross-laboratory study on ceramic electrolytes [25].
Sample Preparation:
- Material Synthesis: Prepare PQD films or solid electrolyte pellets (e.g., LLZO:Ta:Al or LATP) using standardized synthesis protocols to ensure consistent stoichiometry and phase composition [25].
- Electrode Integration: Integrate the sample into an appropriate cell housing, such as a coin cell. Sputter or evaporate ionically-blocking electrodes (e.g., gold or platinum) onto both sides of the pellet to ensure well-defined electrical contact [25].
Data Acquisition:
- Instrument Setup: Use a potentiostat capable of electrochemical impedance spectroscopy. A harmonized testing procedure is recommended to minimize inter-lab deviations [25].
- Measurement Parameters: Apply a small AC perturbation amplitude (e.g., 10-50 mV) to maintain linearity. Sweep the frequency across a wide range, typically from 1 MHz down to 0.1 Hz, at a constant, controlled temperature [25].
- Temperature Control: Conduct measurements in a temperature-controlled environment, allowing sufficient time for temperature equilibration at each step if performing a temperature-dependent study.
Data Analysis:
- Equivalent Circuit Modeling (ECM): Fit the resulting Nyquist plot (imaginary vs. real impedance) with an appropriate equivalent circuit model. A typical model for a solid electrolyte includes a resistor for the bulk ionic conductivity (R_bulk) in series with a constant phase element (CPE) that models the electrode polarization.
- Ionic Conductivity Calculation: Calculate the DC ionic conductivity (σ) using the formula: [ \sigma = \frac{L}{R{bulk} \times A} ] where (L) is the sample thickness, (A) is the electrode area, and (R{bulk}) is the resistance derived from the low-frequency intercept of the impedance arc on the real axis [25].
Protocol: IMS-MS for Surface Ligand Analysis
This protocol describes how to use IMS-MS to analyze the surface ligands of PQDs, which directly influence ionic surface stability.
Sample Preparation:
- PQD Synthesis and Purification: Synthesize PQDs (e.g., CsPbI₃ or FAPbI₃) via hot-injection or ligand-assisted re-precipitation (LARP). Purify the PQDs using antisolvents like methyl acetate to remove excess precursors and ligands [14] [23].
- Ligand Exchange (Optional): To study modified surfaces, perform a post-synthetic ligand exchange. For instance, treat PQDs with a solution of short-chain ligands like 2-aminoethanethiol (AET) in an appropriate solvent, leveraging the strong affinity between thiolate groups and Pb²⁺ on the PQD surface to create a denser passivation layer [23].
- Sample Introduction: Re-dissolve the purified PQD powder in a compatible solvent (e.g., toluene) at a suitable concentration for electrospray ionization (ESI).
Data Acquisition:
- Ionization: Introduce the sample into the IMS-MS system via ESI, carefully optimizing parameters to generate gas-phase ions without degrading the PQDs or their ligand shell.
- Mobility Separation: For DTIMS, apply a uniform, weak electric field (tens of V/cm) across the drift tube filled with an inert buffer gas (e.g., N₂ or He). Pulse ion packets into the drift region and measure their arrival time at the detector [24]. For TWIMS, use a traveling wave device to separate the ions.
- Mass Analysis: After mobility separation, analyze ions by mass-to-charge ratio (m/z) using a time-of-flight (TOF) mass spectrometer.
Data Analysis:
- CCS Calculation (for DTIMS): For each ion species, use the arrival time and experimental parameters (E, P, T, L) in the Mason-Schamp equation to calculate its CCS value [24]. This provides a direct measure of the ion's size, helping to identify different ligand conformations.
- Data Interpretation: Correlate CCS values with m/z signals to identify specific ligand species attached to the PQD surface or released from it. Monitor changes in the IMS-MS spectrum after ligand exchange or stress tests (e.g., heat, light) to track the dissociation of ligands and the formation of new ionic species, which are indicators of surface instability [23].
Visualization of Experimental Workflows
The following diagrams illustrate the logical flow and key components of the experimental techniques discussed.
Ion Mobility Spectrometry Workflow
Ionic Migration Pathways in PQDs
The Scientist's Toolkit: Research Reagent Solutions
Successful experimentation in this field relies on a set of essential materials and reagents. The following table details key items and their specific functions in the context of studying ionic motion in PQDs.
Table 2: Essential Research Reagents and Materials for Ionic Motion Studies
| Category | Specific Example | Function in Experiment |
|---|---|---|
| PQD Core Materials | CsPbBr₃, FAPbI₃ PQDs [14] [23] | The subject of study; their A, B, and X-site ions (e.g., Cs⁺, FA⁺, Pb²⁺, I⁻, Br⁻) undergo migration under stimuli. |
| Surface Ligands | Oleic Acid (OA), Oleylamine (OAm) [23] | Pristine long-chain ligands used in synthesis. Their dynamic binding leads to dissociation and surface defect formation. |
| Ligand Exchange Reagents | 2-Aminoethanethiol (AET) [23] | Used in post-treatment to replace OA/OAm. Strong Pb²⁺-S binding creates a denser, more stable ligand shell, suppressing ion loss. |
| Antisolvents | Methyl Acetate (MeOAc), Methyl Benzoate (MeBz) [14] | Used in purification and interlayer rinsing. Hydrolyze to generate short ligands (e.g., acetate) that can replace pristine long-chain ligands on the PQD surface (X-site). |
| Alkaline Additives | Potassium Hydroxide (KOH) [14] | Added to ester antisolvents to create an alkaline environment that catalyzes hydrolysis, facilitating more efficient ligand exchange. |
| Buffer Gases | Nitrogen (N₂), Helium (He) [24] | Inert gases used in the drift tube of IMS to collide with ions, enabling separation based on mobility. |
| Calibrant Ions | DTIMS-derived CCS calibrants [24] | Ions with known CCS values used to calibrate TWIMS and other secondary IMS methods for accurate CCS determination of unknown analytes. |
The direct observation of ionic motion is paramount to advancing the field of perovskite quantum dots and related ionic materials. Techniques such as IMS-MS, Impedance Spectroscopy, and Precision Laser Spectroscopy provide a multi-faceted toolkit for probing these dynamics across different scales, from bulk film properties to the behavior of single ions and surface ligands. The experimental protocols and reagents detailed in this guide offer a concrete foundation for researchers to implement these methods. By applying these advanced spectroscopic techniques, scientists can uncover the fundamental mechanisms of ionic migration, correlate them with material performance and degradation, and ultimately design next-generation PQDs with enhanced stability and functionality through targeted surface engineering and defect passivation strategies.
In the study of halide perovskite quantum dots (PQDs), understanding and controlling charge transport is paramount for advancing their application in optoelectronic devices and biosensors. The inherent mixed ionic-electronic conductivity of these materials presents a unique challenge, as the interplay between ionic migration and electronic currents directly influences device performance, stability, and efficiency [27]. The ionic nature of the perovskite lattice, characterized by a low activation energy for ion migration, means that under operational stresses such as electric fields, light, or heat, ions can move readily, leading to phenomena like hysteresis, phase segregation, and accelerated degradation [27] [23]. This ionic activity is particularly pronounced at PQD surfaces, where the large surface-to-volume ratio and the prevalence of defects, such as halide vacancies, create pathways for rapid ion transport [23]. Consequently, accurately deconvoluting the ionic and electronic contributions to the overall electrical response is not merely an academic exercise but a critical prerequisite for rational material design, reliable device engineering, and the development of stable PQD-based technologies. This guide provides a detailed framework for achieving this separation, contextualized within the broader research dynamics of ionic migration at PQD surfaces.
Theoretical Foundations of Mixed Conduction
Halide perovskites are classified as mixed ionic-electronic conductors (MIECs), a property stemming from their soft, ionic lattice and defect chemistry. The total current density ((J{total})) flowing through a perovskite material under an applied bias is the sum of the electronic ((J{electronic})) and ionic ((J_{ionic})) components:
[ J{total} = J{electronic} (n, p, \mue, \muh) + J{ionic} (ci, \mu_i) ]
Here, the electronic contribution depends on the concentration and mobility of electrons ((n, \mue)) and holes ((p, \muh)), while the ionic contribution depends on the concentration and mobility of mobile ions ((ci, \mui)), such as halide anions (I⁻, Br⁻, Cl⁻), A-site cations (Cs⁺, MA⁺, FA⁺), and Pb²⁺ cations [27]. The low formation energy for defects, especially halide vacancies, facilitates ion migration with activation energies often ranging between 0.1 and 0.5 eV, making it a dominant process at room temperature [23].
Ionic migration in PQDs is not a bulk phenomenon alone; it is profoundly influenced by surface chemistry. The detachment of surface ligands during synthesis or purification creates coordinatively unsaturated sites (e.g., under-coordinated Pb²⁺ ions), which act as traps for charge carriers and nucleation points for ionic defects [23] [14]. Furthermore, the dissociation of weakly bound ligands like oleic acid and oleylamine increases surface disorder, accelerating halide migration and subsequent structural degradation [23]. Therefore, any electrical characterization must account for this surface-mediated ionic transport, which can dominate the overall electrical response in quantum-confined systems.
Table 1: Key Mobile Ions in Halide Perovskites and Their Characteristics
| Ion Type | Specific Ions | Typical Activation Energy (eV) | Primary Impact on Device Properties |
|---|---|---|---|
| Halide Anions | I⁻, Br⁻, Cl⁻ | 0.1 - 0.5 | Phase segregation, hysteresis, trap state formation [27] [23] |
| A-site Cations | Cs⁺, MA⁺, FA⁺ | >0.5 | Phase stability, ionic polarization [27] |
| B-site Cations | Pb²⁺ | >0.5 | Non-radiative recombination, structural degradation [27] |
| Extrinsic Ions | H⁺, O²⁻, Na⁺ | Varies | Doping, accelerated aging, chemical degradation [27] |
Experimental Techniques for Deconvolution
Separating the ionic and electronic components requires a combination of techniques, each probing different aspects of the charge transport phenomenon. Environmental control is critical during all measurements, as perovskites are highly sensitive to external stimuli like humidity, light, electric field, and heat [27].
Direct Current (DC) Polarization with Blocking Electrodes
This method is a cornerstone for quantifying ionic conductivity. It employs electronically blocking electrodes (e.g., Au, C) that prevent steady-state electronic current, forcing the measured long-time-scale current to be predominantly ionic.
Experimental Protocol:
- Device Fabrication: Deposit a PQD thin film onto an insulating substrate (e.g., glass, SiO₂/Si). Pattern two metal electrodes (e.g., Au, 50-100 nm thick) with a well-defined gap (e.g., 10-100 µm) on top of the film.
- Measurement Setup: Place the device in a controlled environment (e.g., inert gas glovebox, vacuum probe station). Connect the electrodes to a source measure unit (SMU) or a high-resolution electrometer.
- Voltage Application: Apply a constant DC voltage bias (e.g., 0.1 - 1 V) across the electrodes. The voltage must be kept low to avoid irreversible electrochemical reactions.
- Current Transient Measurement: Record the current transient over time. The current will initially be high due to the displacement of all charge carriers (electronic and ionic) but will decay as the mobile ions are blocked at the electrodes, building up a counteracting space-charge field.
- Data Analysis: The steady-state current ((I{SS})) is attributed to the residual electronic leakage. The initial current ((I0)) contains both ionic and electronic contributions. The ionic conductivity ((\sigma{ionic})) can be extracted using the equation: [ \sigma{ionic} = \frac{I0 - I{SS}}{V} \cdot \frac{d}{w \cdot t} ] where (V) is the applied voltage, (d) is the electrode gap, (w) is the electrode width, and (t) is the film thickness.
Electrochemical Impedance Spectroscopy (EIS)
EIS is a powerful technique for probing different relaxation processes in a material by measuring its impedance as a function of frequency.
Experimental Protocol:
- Cell Configuration: Use a symmetric cell structure with either blocking (e.g., Au/PQD/Au) or non-blocking (e.g., ITO/PQD/Ag) electrodes, depending on the target information.
- Measurement: Apply a small AC perturbation signal (10-50 mV) superimposed on a DC bias, sweeping across a wide frequency range (e.g., 1 MHz to 0.1 Hz).
- Data Fitting and Analysis: The resulting Nyquist plot (imaginary vs. real impedance) is fitted to an equivalent circuit model. A common model for perovskites is (RₑₗRᵢₒₙCPEᵢₒₙ), where:
- (Rₑₗ) represents the electronic resistance.
- (Rᵢₒₙ) represents the ionic transport resistance.
- (CPEᵢₒₙ) is a constant phase element modeling the capacitive behavior associated with ionic diffusion and electrode polarization. The ionic conductivity is calculated from (Rᵢₒₙ) using the sample geometry. The characteristic frequency of the ionic arc can provide information about ion migration timescales [27].
Field-Effect Transistor (FET) Measurements
FET configurations can be used to modulate electronic carrier density while monitoring transport, helping to isolate electronic mobility from ionic effects.
Experimental Protocol:
- Transistor Fabrication: Fabricate a bottom-gate top-contact FET structure. A heavily doped Si wafer with a thermal SiO₂ layer serves as the gate and gate dielectric, respectively. The PQD film is spin-coated onto the SiO₂, and source/drain electrodes (e.g., Au) are deposited on top.
- Transfer Characteristic Measurement: In a shielded, inert environment, sweep the gate voltage ((VG)) while keeping a small source-drain voltage ((V{DS})) constant. Measure the source-drain current ((I_{DS})).
- Hysteresis Analysis: The hysteresis between forward and reverse (V_G) sweeps is a direct signature of ionic motion. Mobile ions migrate to screen the gate field, effectively doping the channel and shifting the threshold voltage.
- Electronic Mobility Extraction: The field-effect mobility of electrons or holes is extracted from the linear or saturation regime of the (I{DS})-(VG) curve in the sweep direction with minimal hysteresis, providing a measure of electronic transport decoupled from slow ionic processes.
Diagram 1: Experimental workflow for separating ionic and electronic contributions.
Advanced Quantification and Modeling Tools
Beyond basic characterization, advanced toolsets are required to quantify the kinetics and impact of ionic migration.
Theoretical Modeling:
- Density Functional Theory (DFT): Used to calculate activation energies for the migration of various ions by determining the energy barrier for an ion to move from one lattice site to a neighboring vacancy [27]. This helps predict which ions are most mobile.
- Drift-Diffusion Models: Numerical simulations that solve coupled equations for electronic and ionic charge transport, incorporating drift in the electric field and diffusion due to concentration gradients. These models can fit experimental current-voltage (I-V) data to extract carrier mobilities and ion diffusion coefficients.
Elemental and Molecular Analysis:
- X-ray Photoelectron Spectroscopy (XPS): Can detect changes in surface stoichiometry after electrical biasing, providing direct evidence of ion migration (e.g., halide depletion at the anode) [27].
- Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS): Offers high-sensitivity depth profiling to track the movement of specific isotopes or ions across a device, providing a direct visualization of ionic migration [27].
Table 2: Comparison of Key Characterization Techniques
| Technique | Primary Output | Information on Ionic Transport | Information on Electronic Transport | Key Limitations |
|---|---|---|---|---|
| DC Polarization | Current transient | Ionic conductivity, diffusion coefficient | Electronic leakage current | Requires perfectly blocking electrodes; long measurement times [27] |
| Impedance Spectroscopy | Complex impedance | Ionic resistance, capacitance, diffusion timescales | Electronic (bulk) resistance | Complex data interpretation; requires accurate equivalent circuit model [27] |
| FET Measurement | Transfer curve (ID-VG) | Hysteresis, ion migration timescales | Field-effect mobility, carrier type | PQD film quality and electrode contacts critically affect results [23] |
| Drift-Diffusion Modeling | Simulated I-V curves | Fitted ion mobility, concentration | Fitted electron/hole mobility | Relies on input parameters; can be computationally intensive [27] |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials and reagents essential for experiments focused on the electrical characterization and stabilization of PQD surfaces.
Table 3: Research Reagent Solutions for PQD Surface and Conductivity Studies
| Item/Chemical | Function/Application | Technical Notes |
|---|---|---|
| Methyl Benzoate (MeBz) | Ester antisolvent for interlayer rinsing of PQD films. Hydrolyzes to conductive benzoate ligands that replace insulating oleate [14]. | Preferred over methyl acetate due to stronger binding of hydrolyzed ligand; used in Alkali-Augmented Antisolvent Hydrolysis (AAAH) [14]. |
| Potassium Hydroxide (KOH) | Alkaline additive to ester antisolvents. Catalyzes ester hydrolysis, enabling rapid and dense conductive capping on PQD surfaces [14]. | Lowers hydrolysis activation energy ~9-fold; critical for the AAAH strategy to improve surface conductivity and reduce defects [14]. |
| 2-Aminoethanethiol (AET) | Short-chain ligand for post-synthetic surface passivation. Binds strongly to under-coordinated Pb²⁺ sites via thiol group [23]. | Heals surface defects, inhibits ligand dissociation, and improves stability against moisture and UV light [23]. |
| C18 Reversed-Phase Silica Beads | Chromatographic substrate in µSIC sensors. Binds hydrophobic analytes, altering surface ionic conductivity for detection [28]. | Enables label-free, non-optical sensing of molecules via changes in surface charge and ionic conduction [28]. |
| Deep Eutectic Solvent (DES) | Solvent for conductive gels. Comprises glycerol and choline chloride; provides ionic conductivity and enhances mechanical properties [29]. | Used in biogels for epidermal electronics; offers high signal-to-noise ratio for physiological signal acquisition [29]. |
| PEDOT:PSS | Conductive polymer for semi-interpenetrating networks. Provides electronic conduction pathway in composite gels and films [29]. | Enhances both mechanical strength and electrical conductivity in soft electronic interfaces [29]. |
Application in Biosensing: A Case Study on μSIC
The practical importance of surface ionic conduction is exemplified by the microscale surface ion conduction (μSIC) sensor, a platform for detecting biological analytes and small molecules [28]. This sensor directly leverages changes in surface ionic conductivity for transduction.
Working Principle: The μSIC sensor features a microfluidic channel packed with a bed of functionalized microbeads (e.g., C18 silica). The surface of these beads carries a charge that governs ionic transport within the electrical double layer (EDL). When a target analyte (e.g., a negatively charged PFAS molecule) binds to the bead surface, it alters the local surface charge density and zeta potential. This change modulates the ionic conductivity along the bead surface, which is measured as a shift in the transverse current flowing through the bed upon application of a small AC or DC voltage [28].
Experimental Protocol for PFAS Detection:
- Sensor Fabrication: A polydimethylsiloxane (PDMS) microchannel is fabricated using soft lithography. It is packed with two adjacent beds: a primary bed of silver-coated glass beads (3D ground electrode) and a secondary bed of C18 chromatographic beads as the sensing zone.
- Sample Introduction: An aqueous sample containing the target PFAS (e.g., perfluorooctane sulfonic acid, PFOS) in a background electrolyte (e.g., 1 mM KCl) is flowed through the channel.
- Measurement: A voltage (e.g., 1 V) is applied between the inlet/outlet and the 3D ground electrode. The current is monitored over time.
- Signal Enhancement (Optional): To boost sensitivity for low-abundance analytes, faradaic ion concentration polarization (fICP) can be integrated. Applying a potential bias generates an ion depletion zone (IDZ), which creates a steep electric field gradient. This gradient acts to preconcentrate the charged analytes at the C18 bead bed via counter-flow focusing, increasing the binding density and the resultant current shift by orders of magnitude [28].
- Data Analysis: The change in current ((\Delta I)) is correlated with the logarithm of the analyte concentration. A calibration curve is used for quantification.
Diagram 2: Signaling logic of a μSIC sensor based on surface ion conduction.
The separation of ionic and electronic contributions is a fundamental and non-negotiable aspect of advanced electrical characterization in halide perovskite research. The methodologies outlined herein—from foundational DC and EIS techniques to advanced modeling and sensor applications—provide a comprehensive toolkit for researchers to dissect the complex charge transport landscape in PQDs. Mastering this deconvolution is the key to unlocking a deeper understanding of ionic migration dynamics at PQD surfaces, which in turn enables the rational design of more stable, efficient, and reliable perovskite-based optoelectronics and biosensors. As the field progresses, the development of standardized protocols and in-situ characterization methods will further refine our ability to observe and control these intertwined phenomena.
The stability and functional performance of Perovskite Quantum Dots (PQDs) are intrinsically governed by ionic migration dynamics at their surface. The inherent ionic crystal lattice of lead halide perovskites, while granting exceptional optoelectronic properties, also facilitates the rapid migration of ions, leading to material degradation and performance decay. Ligand engineering addresses this fundamental challenge by creating designed surface capping layers that function as dynamic "ionic gates." These engineered layers selectively control the ingress and egress of ionic species, thereby stabilizing the perovskite crystal structure against environmental factors such as moisture, oxygen, and light, while simultaneously fine-tuning its electronic properties for applications ranging from biosensing to photovoltaics.
The susceptibility of PQDs to polar solvents, moisture, and light primarily stems from highly ionic and dynamic interactions between surface ligands and the PQD core, which cause rapid ligand desorption and damage crystal integrity [30]. Furthermore, the low formation energy of the crystal lattice and high delocalization activity of surface ions exacerbate this instability [30]. The capping layer thus serves not merely as a passive barrier but as an active interfacial region that modulates ionic traffic, repels detrimental species, and passivates electronic traps, making ligand engineering a cornerstone for advancing PQD technologies.
Quantitative Analysis of Ligand Engineering Strategies
The effectiveness of various ligand engineering strategies can be quantitatively assessed through key performance metrics, including photoluminescence (PL) enhancement, stability improvement, and device efficiency. The following table synthesizes experimental data from recent studies on CsPbBr₃ PQDs and solar cells, providing a comparative overview of different approaches.
Table 1: Quantitative Performance of Ligand Engineering and Surface Capping Strategies
| Material/System | Engineering Strategy | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| CsPbBr₃ PQD Film | Sequential treatment: MeOAc (ligand removal), CsAc (defect repair), HMDS (SiOₓ encapsulation) | PL Intensity Enhancement | 3.4x increase vs. untreated film | [30] |
| CsPbBr₃ PQD Film | Sequential treatment: MeOAc, CsAc, HMDS | Ambient Stability Improvement | ~40% reduced instability vs. untreated film | [30] |
| FAPbI₃ Solar Cell | BAAc (n-butylamine acetate) ionic liquid capping layer | Power Conversion Efficiency (PCE) | 24.76% (vs. 20.53% for untreated device) | [31] |
| FAPbI₃ Solar Cell | BAAc ionic liquid capping layer | Open-Circuit Voltage (Voc) | 1.19 V (vs. 1.08 V for untreated device) | [31] |
| FAPbI₃ Solar Cell (Mini-Module) | BAAc ionic liquid capping layer | PCE on Active Area | 20.47% (vs. 17.39% for untreated module) | [31] |
| FAPbI₃ Solar Cell | BAAc ionic liquid capping layer | Long-Term Stability (T80, unencapsulated) | 3500 hours (at 35% relative humidity) | [31] |
The data underscores the profound impact of synergistic surface treatments. The sequential application of ligand removal, ionic defect repair, and robust encapsulation yields the most significant improvements in both optical performance and environmental resilience [30]. Similarly, the use of advanced ionic liquid cappers like BAAc demonstrates that effective surface engineering directly translates to superior device-level performance and remarkable operational longevity [31].
Experimental Protocols for Surface Capping and Analysis
Protocol: Sequential Post-Treatment of CsPbBr₃ PQD Films
This protocol details the synergistic process for enhancing the brightness and stability of CsPbBr₃ PQD films, as validated by Zhang et al. [30].
Step 1: Ligand Removal via Methyl Acetate (MeOAc) Washing
- Procedure: Spin-coat the as-synthesized CsPbBr₃ PQD film. Subsequently, wash the film by dynamically dripping methyl acetate (MeOAc) onto the spinning substrate for 20 seconds.
- Rationale: MeOAc effectively removes long-chain insulating ligands like oleic acid and oleylamine without dissolving the PQD core. This is confirmed by Fourier-Transform Infrared (FTIR) spectroscopy, showing significant reduction of peaks at 2853 cm⁻¹ and 2924 cm⁻¹ (C-H stretching) and 1710 cm⁻¹ (C=O stretching) [30].
- Critical Note: Ligand removal is a prerequisite for effective subsequent defect repair and encapsulation.
Step 2: Defect Repair via Cesium Acetate (CsAc) Treatment
- Procedure: After MeOAc washing, immediately deposit a solution of Cesium Acetate (CsAc) in isopropanol (concentration: 2 mg/mL) onto the film via spin-coating.
- Rationale: The Cs⁺ ions from CsAc preferentially fill cesium vacancies (VˍCs) on the PQD surface. This repair of cationic vacancies reduces non-radiative recombination centers, which is a primary mechanism for the observed boost in photoluminescence intensity [30].
Step 3: Surface Encapsulation via HMDS-derived SiOˍx Passivation
- Procedure: After CsAc treatment, coat the film with Hexamethyldisilazane (HMDS) by spin-coating. The HMDS layer undergoes hydrolysis in ambient moisture, forming a thin, protective silicon oxide (SiOˍx) layer on the PQD surface.
- Rationale: The SiOˍx layer acts as a robust physical barrier against moisture and oxygen, significantly enhancing the film's ambient stability without compromising its optical properties [30].
Diagram: Workflow for Sequential Post-Treatment of PQD Films
Protocol: Ionic Liquid Capping of FAPbI₃ for Photovoltaics
This protocol describes the application of a room-temperature ionic liquid (IL) capping layer for high-performance perovskite solar cells, based on the work with n-butylamine acetate (BAAc) [31].
Step 1: Perovskite Film Preparation and Pre-crystallization
- Procedure: Deposit the FAPbI₃ perovskite precursor solution (in DMF/DMSO solvent mix) using a one-step spin-coating method. Allow the film to pre-crystallize.
- Rationale: This forms the underlying 3D perovskite absorber layer.
Step 2: Room-Temperature Ionic Liquid Capping
- Procedure: Without any thermal treatment, spin-coat an isopropyl alcohol (IPA) solution of n-butylamine acetate (BAAc) directly onto the pre-crystallized FAPbI₃ film. This is denoted as the BAAc RT method.
- Rationale: The BAAc IL provides uniform and thorough passivation of surface defects. The acetate anion (Ac⁻) offers better energy level alignment for hole extraction compared to halide anions like I⁻ [31]. Crucially, this room-temperature process avoids the formation of a 2D perovskite phase that can occur with thermal annealing (BAAc TA), which often impedes charge transport due to quantum confinement effects [31].
Step 3: Device Fabrication and Characterization
- Procedure: Complete the fabrication of the n–i–p solar cell stack. Characterize using current-density voltage (J-V) measurements for PCE and Voc, and conduct long-term stability tests under controlled humidity.
- Key Analysis: Grazing-incidence wide-angle X-ray scattering (GIWAXS) should be used to confirm the absence of a 2D phase in the BAAc RT sample and to assess crystal orientation. Time-resolved photoluminescence (TRPL) can quantify the reduction in non-radiative recombination [31].
The Scientist's Toolkit: Essential Reagents for Ligand Engineering
Successful implementation of ligand engineering strategies requires a curated set of chemical reagents and analytical tools. The following table itemizes key materials and their specific functions in the development of advanced capping layers.
Table 2: Research Reagent Solutions for Ligand Engineering
| Reagent / Material | Function in Ligand Engineering | Technical Notes |
|---|---|---|
| Methyl Acetate (MeOAc) | Solvent Wash: Selectively removes long-chain native ligands (OA, OAm) without damaging PQD core. | Preferable to non-polar solvents for more complete ligand removal, crucial for subsequent treatment efficacy [30]. |
| Cesium Acetate (CsAc) | Defect Repair Agent: Source of Cs⁺ ions to fill cationic vacancies (VˍCs) on the PQD surface. | Applied in isopropanol solution post-ligand removal. Directly reduces trap states, boosting PL [30]. |
| Hexamethyldisilazane (HMDS) | Precursor for Encapsulation: Hydrolyzes to form a protective silicon oxide (SiOˍx) layer upon air exposure. | Provides a moisture-resistant barrier, significantly enhancing ambient stability of the film [30]. |
| n-Butylamine Acetate (BAAc) | Ionic Liquid Capping Agent: Passivates surface defects and improves energy level alignment. | Room-temperature processing avoids formation of charge-blocking 2D phases. Enables high Voc in solar cells [31]. |
| Oleic Acid (OA) / Oleylamine (OAm) | Native Ligands: Used during synthesis for colloidal stability and size control. | Typically removed or exchanged post-synthesis to improve inter-dot charge transport in devices [30]. |
Molecular Dynamics and Future Perspectives
The design of capping layers is increasingly informed by molecular-level simulations. Molecular dynamics (MD) simulations provide nanoscale insights into how ionic environments and surface morphology influence adsorption and nucleation processes. For instance, studies on biphasic calcium phosphate surfaces reveal that ions like PO₄³⁻ and HPO₄²⁻ are effectively adsorbed, with a preference for grain boundary regions, and that introduced nanogrooves—particularly square grooves—significantly increase ion adsorption capacity compared to planar surfaces [32]. These principles are translatable to PQD systems, suggesting that engineering specific surface topographies and chemical environments at the nanoscale can optimize the passivation and stability conferred by capping layers.
Future research will focus on integrating these insights to create multi-functional, smart ionic gates. Key directions include the development of lead-free perovskite capping layers (e.g., based on bismuth compositions like Cs₃Bi₂Br₉) to address toxicity concerns [13], and the fusion of ligand engineering with machine learning for the high-throughput design of next-generation capping molecules. The ultimate goal is the rational creation of dynamic capping layers that can selectively gate ionic fluxes in response to environmental stimuli, paving the way for PQD-based devices with unparalleled performance and operational lifetime.
Diagram: Ionic Gating Mechanism of a Surface Capping Layer
Strategic Ion Doping (e.g., Ag+) to Modulate Migration Barriers and Enhance Photoluminescence
Ionic migration within halide perovskite quantum dots (PQDs) represents a fundamental challenge that impedes their commercial application in optoelectronic devices. This ionic mobility, particularly of halide ions, leads to phase segregation, defect formation, and subsequent degradation of photoluminescence (PL) performance [23] [33]. Within the broader context of ionic migration dynamics in PQD surfaces research, strategic ion doping has emerged as a powerful technique to modulate migration barriers and enhance optoelectronic properties. This technical guide comprehensively examines the mechanisms, methodologies, and outcomes of Ag+ doping as a strategic intervention for improving the structural and photoluminescence stability of PQDs, providing researchers with both theoretical foundations and practical experimental protocols.
The Problem: Ionic Migration and Structural Instability in PQDs
The exceptional optoelectronic properties of PQDs are counterbalanced by their inherent ionic nature, which facilitates ion migration under external stimuli such as light, heat, and electric fields. The structural degradation of PQDs primarily occurs through two interconnected mechanisms:
- Defect Formation via Surface Ligand Dissociation: Weakly bound surface ligands (e.g., oleic acid and oleylamine) readily detach from PQD surfaces, creating unsaturated coordination sites and surface defects that accelerate degradation [23].
- Vacancy-Driven Halide Migration: The low formation energy of halide vacancies within the PQD lattice enables facile ion migration, leading to non-stoichiometric compositions and phase segregation, particularly in mixed-halide perovskites [23] [33].
These degradation pathways manifest experimentally as color deterioration and intensity decline in photoluminescence, severely limiting the practical application of PQDs in light-emitting devices [33]. The ionic migration energy barriers, which determine the kinetics of these processes, thus become critical parameters for controlling PQD stability.
Strategic Ion Doping: Mechanisms and Energetics
Strategic incorporation of heterovalent ions, particularly Ag+, into the PQD lattice modifies the fundamental energetics of ionic migration through multiple concurrent mechanisms:
Bandgap Engineering and Electronic Structure Modification
Density functional theory (DFT) calculations demonstrate that Ag+ doping induces a widening of the PQD bandgap, which contributes to reduced non-radiative recombination and enhanced photoluminescence quantum yield (PLQY) [34]. This bandgap modification alters the charge carrier dynamics, favoring radiative recombination pathways.
Localized Surface Plasmon Resonance (LSPR) Enhancement
At optimal doping concentrations (0.4 mol% AgI), Ag nanoparticles form within the glass matrix, generating a localized surface plasmon resonance effect under visible light excitation [34]. This LSPR effect produces a strongly enhanced electromagnetic near-field that increases charge carrier density and optical activity, thereby boosting PL intensity.
Lattice Stabilization and Vacancy Reduction
Ag+ incorporation stabilizes the PQD crystal lattice by reducing structural stress and lattice mismatch, ultimately minimizing halide vacancy formation [34]. The presence of Ag+ ions further induces a charge compensation effect that enhances the migration and rearrangement of Cs+ and Pb2+ ions, promoting more thermodynamically stable crystalline structures [34].
Table 1: Quantitative Enhancement in Photoluminescence Properties through Ag+ Doping
| Material System | Doping Concentration | PLQY Enhancement | Stability Improvement | Key Mechanisms |
|---|---|---|---|---|
| CsPbBrI₂ PQD Glass | 0.4 mol% AgI | 20% → 62.4% | Maintains >88% initial emission under prolonged stress | LSPR, Bandgap widening, Lattice stabilization |
| CsPbBr₃ PQDs | Not specified | Significant PLQY enhancement through LSPR | Improved thermal and photostability | LSPR effect from Ag nanoparticles |
| AgInS₂ QDs | Cd²⁺ doping | Increased PL intensity | - | Reduced surface-to-volume ratio, Surface structure modification |
Experimental Protocols: AgI Doping in CsPbBrI₂ PQD Glass
Materials and Synthesis Methodology
Reagents:
- Glass precursors: SiO₂, B₂O₃, ZnO, Na₂CO₃ (purity: 99.99%)
- Perovskite precursors: Cs₂CO₃, PbBr₂, NaBr, PbI₂, NaI (purity: 99%)
- Dopant source: AgI (purity: 99%)
- All chemical reagents available from Aladdin
Synthesis Procedure:
Batch Preparation: Weigh a total mass of 10g of raw materials according to the molar composition: 86 mol% (SiO₂-B₂O₃-ZnO-Na₂CO₃) and 14 mol% (Cs₂CO₃-PbBr₂-NaBr-PbI₂-NaI), with additional X mol% AgI (where X = 0, 0.1, 0.2, 0.4, and 0.6 mol%) [34].
Homogenization: Thoroughly grind the mixture in an agate mortar for 45 minutes to ensure uniform mixing of precursors [34].
Melting and Quenching: Transfer the well-ground materials to an alumina crucible and melt in a high-temperature furnace at 1350°C for 30 minutes. Subsequently, pour the melt onto a preheated copper mold and press with another plate to form glass samples of uniform thickness [34].
Annealing: Immediately transfer the quenched glass to a muffle furnace preheated to 450°C for annealing over 4 hours to relieve internal stresses, then gradually cool to room temperature [34].
Controlled Crystallization: Subject the annealed glass samples to a two-step heat treatment process: first at the glass transition temperature (Tg) for nucleation, followed by heating at the perovskite crystallization temperature (Tc) for grain growth, facilitating the simultaneous precipitation of CsPbBrI₂ PQDs and Ag nanoparticles within the glass matrix [34].
Characterization and Validation Techniques
- Structural Analysis: X-ray diffraction (XRD) to monitor crystallization progress and phase purity [34]
- Optical Properties: UV-Vis absorption spectroscopy and photoluminescence spectroscopy to quantify bandgap changes and emission characteristics [34]
- Performance Metrics: Photoluminescence quantum yield (PLQY) measurements to quantify enhancement efficiency [34]
- Stability Assessment: Thermal, water, and photostability tests under prolonged stress conditions [34]
Complementary Stabilization Approaches
While Ag+ doping addresses intrinsic lattice instability, combined strategies often yield superior results:
Surface Ligand Engineering
Surface modification with zwitterionic ligands (e.g., sulfobetaine-18) retards ion migration and accelerates entropy-driven reverse processes in mixed-halide PQDs, enabling unprecedented PL stability with negligible wavelength shifts (<3 nm) during prolonged photoexcitation [33]. Alternative ligand systems including trioctylphosphine (TOP), trioctylphosphine oxide (TOPO), and L-phenylalanine (L-PHE) effectively suppress non-radiative recombination by coordinating with undercoordinated Pb²⁺ ions and surface defects, with PL intensity enhancements of 3-18% reported [35].
Core-Shell Structures and Crosslinking
Overcoating PQDs with wider bandgap semiconductors (e.g., ZnS shell on AgInS₂ core) significantly enhances PL intensity by reducing surface defects and suppressing non-radiative recombination pathways [36] [37]. Similarly, introducing crosslinkable ligands that form interconnected networks upon light or heat exposure minimizes ligand dissociation and subsequent defect formation [23].
Table 2: Comparison of PQD Stabilization Strategies
| Strategy | Primary Mechanism | Key Advantages | Limitations | Reported Efficacy |
|---|---|---|---|---|
| Ag⁺ Doping | Lattice stabilization, Bandgap modification, LSPR | Addresses intrinsic lattice instability, Enhances charge carrier density | Optimal concentration critical (0.4 mol%) | PLQY: 20% → 62.4% [34] |
| Ligand Engineering | Surface defect passivation, Ion migration suppression | Tunable chemical functionality, Applicable to various PQD systems | Potential conductivity reduction | PL retention: >70% after 20 days UV [35] |
| Core-Shell Structures | Spatial carrier confinement, Surface defect reduction | Excellent environmental stability, Broad material selection | Interface lattice mismatch, Synthesis complexity | Highest PL intensity for AgInS₂:Cd²⁺/ZnS [37] |
| Crosslinking | Mechanical stabilization, Ligand dissociation prevention | Enhanced structural integrity, Improved processability | Potential impact on quantum confinement | Improved structural stability [23] |
The Researcher's Toolkit: Essential Materials and Reagents
Table 3: Essential Research Reagents for Ag⁺ Doping Experiments
| Reagent Category | Specific Examples | Function/Purpose | Technical Considerations |
|---|---|---|---|
| Glass Matrix Precursors | SiO₂, B₂O₃, ZnO, Na₂CO₃ (99.99%) | Forms stable, amorphous host matrix | High purity critical for reproducible optical properties [34] |
| Perovskite Precursors | Cs₂CO₃, PbBr₂, PbI₂, NaBr, NaI | Sources of PQD constituent ions | Stoichiometric ratios determine halide composition [34] |
| Dopant Sources | AgI (99%) | Provides Ag⁺ ions for strategic doping | Concentration optimization essential (0.1-0.6 mol%) [34] |
| Surface Ligands | Oleic Acid (OA), Oleylamine (OAm) | Controls nanocrystal growth and dispersion | Bent chain structure causes steric hindrance [23] |
| Alternative Ligands | TOP, TOPO, L-PHE, Sulfobetaine-18 | Enhanced surface passivation and defect suppression | Specific coordination mechanisms with Pb²⁺ sites [35] [33] |
| Solvents | Chloroform, Toluene, Chlorobenzene | Dispersion medium for QDs and processing | Polarity affects ligand binding and colloidal stability [38] |
Strategic Ag+ doping represents a multifaceted approach to simultaneously modulate ionic migration barriers and enhance photoluminescence in PQDs. Through the combined mechanisms of LSPR enhancement, bandgap engineering, and lattice stabilization, researchers can achieve remarkable improvements in both PLQY (up to 62.4%) and operational stability (>88% emission retention). The experimental protocols outlined herein provide a reproducible methodology for implementing this approach in glass-embedded PQD systems.
Future research directions should focus on optimizing doping concentrations across different PQD compositions, exploring combinatorial approaches with surface ligand engineering, and developing advanced characterization techniques to directly probe the modified ion migration pathways. As the field progresses toward practical applications, understanding the synergistic effects of multiple stabilization strategies will be crucial for designing next-generation PQD materials with exceptional performance and durability.
Ionic migration dynamics, a phenomenon extensively studied in materials science such as in perovskite quantum dots (PQDs) where ion movement affects stability and performance, find a parallel and transformative application in the field of transdermal drug delivery [9] [39]. In PQDs, the controlled manipulation of ionic movement, including the use of cation additives and surface ligand engineering to suppress halide vacancy migration, is key to enhancing material stability [39]. Translating this principle, ionic liquids (ILs)—organic salts liquid at room temperature—are engineered to dynamically interact with and modulate the skin's barrier properties. These tunable materials, composed of various cation and anion pairs, function as powerful permeation enhancers by disrupting the densely packed lipid matrix of the stratum corneum (SC), the outermost skin layer [40]. This approach is particularly vital for delivering biopharmaceuticals, including proteins, peptides, and nucleic acids, which cannot passively cross this formidable barrier [40] [41]. By acting simultaneously as solvents and permeation enhancers, IL-based platforms can significantly improve the solubility and stability of labile biomolecules, heralding a new era for non-invasive delivery of therapeutics ranging from insulin for diabetes to antigens for transdermal vaccination [40] [42].
Fundamentals of Ionic Liquids in Transdermal Delivery
Composition and Design Principles
Ionic liquids are defined by their tunable cations and anions, which dictate their physicochemical properties and biological interactions. For transdermal applications, the move toward biocompatible ILs is paramount. These are increasingly formed from safe, endogenous materials such as choline as cations, and amino acids, fatty acids, or lipids as anions [41] [42]. For instance, one commonly studied biocompatible IL uses 1,2-dimyristoyl-sn-glycero-3 ethyl-phosphocholine (EDMPC) as the cation and linoleic acid (Lin) as the anion, forming [EDMPC][Lin] [42]. The mechanism of action for ILs as permeation enhancers involves a multi-pronged approach:
- Lipid Disruption: ILs can interact with and fluidize the structured intercellular lipids in the SC, reducing their barrier function [40].
- Protein Interaction: Some ILs can alter the conformation of keratin within corneocytes, further facilitating permeation [40].
- Solubilization Power: ILs can dramatically improve the solubility of both hydrophilic and hydrophobic drugs, increasing the thermodynamic driving force for skin penetration [43] [41].
Comparative Analysis of Common IL Formulations
The versatility of ILs allows for their integration into a variety of advanced formulation architectures, each offering distinct advantages for specific delivery challenges. The table below summarizes the key characteristics of predominant IL-based platforms.
Table 1: Overview of Key Ionic Liquid-Based Formulations for Transdermal Delivery
| Formulation Type | Key Composition | Mechanism of Action | Advantages | Representative Therapeutics Delivered |
|---|---|---|---|---|
| IL-based Nanoemulsions (e.g., IL-in-Oil) | ILs dispersed in oil phase (e.g., Isopropyl Myristate) stabilized with surfactants [40] [44] | Forms nanoscale droplets; IL disrupts SC, oil phase modulates drug partitioning | Enhanced drug stability, improved skin permeation of hydrophobic drugs [40] | Dacarbazine derivatives for melanoma therapy [44] |
| IL Self-Assembled Micelles (ILs-SAMs) | ILs forming micellar structures through intermolecular interactions in aqueous media [43] | Nanocarriers for solubilizing drugs; IL components enhance permeation | High stability, targeted delivery, controllable release profile [43] | Paclitaxel (skin cancer), antifungal agents [43] |
| Solid-in-Oil (S/O) Nanodispersions | Drug particles nanodispersed in oil phase with ILs [40] [42] | ILs coat drug nanoparticles, facilitating transport through lipidic SC | Superior delivery of hydrophilic macromolecules and vaccines [42] | Ovalbumin (antigen), insulin [40] [42] |
| IL-loaded Patches | IL formulation (e.g., S/O) incorporated into a Pressure-Sensitive Adhesive (PSA) [42] | PSA provides intimate skin contact; IL formulation enables sustained release | Excellent skin retention, controlled release, patient compliance [42] | Acyclovir, Ovalbumin (transdermal vaccine) [42] |
Key Experimental Protocols and Methodologies
Protocol 1: Formulation of an IL-based Solid-in-Oil (S/O) Nanodispersion
This protocol is foundational for creating formulations suitable for delivering macromolecular drugs like proteins and antigens [42].
Materials:
- Ionic Liquid: Biocompatible [EDMPC][Lin] IL.
- Oil Phase: Isopropyl Myristate (IPM).
- Aqueous Drug Solution: Contains the active pharmaceutical ingredient (e.g., Ovalbumin, Insulin).
- Surfactant: Such as sucrose laurate.
Procedure:
- Primary Water-in-Oil (W/O) Emulsion: The aqueous drug solution is added to a mixture of IPM and surfactant. This system is emulsified using a high-speed homogenizer (e.g., 12,000 rpm for 2 minutes) to form a coarse W/O emulsion.
- Secondary Emulsification: The coarse emulsion is further subjected to probe sonication on an ice bath (e.g., 40-50 W output for 2 minutes) to form a fine and stable W/O emulsion.
- Removal of Water: The fine W/O emulsion is placed under a vacuum (e.g., 0.1 MPa) at a controlled temperature (e.g., 40°C) for at least 2 hours to completely evaporate the dispersed aqueous phase. This process results in a solid-in-oil (S/O) nanodispersion where the drug nanoparticles are coated with the IL and surfactant, suspended in the continuous oil phase (IPM).
Protocol 2: Fabrication of an IL-based Transdermal Patch
This protocol details the conversion of a liquid IL formulation into a solid patch for convenient and prolonged application [42].
Materials:
- IL-S/O Nanodispersion: As prepared in Protocol 1.
- Pressure-Sensitive Adhesive (PSA): e.g., DURO-TAK 87-4098.
- Patch Components: Release liner and backing (support) film.
Procedure:
- Mixing: The IL-S/O nanodispersion is mixed with an equal weight (1:1 ratio) of the selected PSA. The mixture is stirred thoroughly until a homogeneous blend is achieved.
- Coating and Drying: The resulting adhesive mixture is coated onto a release liner using a controlled method (e.g., an applicator) to achieve a uniform thickness (e.g., 100 µm).
- Drying and Lamination: The coated liner is transferred to an oven to evaporate any residual volatile solvents. Subsequently, a backing film is laminated onto the dried adhesive layer.
- Punching and Storage: The final laminate is punched into patches of the desired size and stored under controlled conditions.
Diagram: Workflow for IL-based Transdermal Patch Fabrication
Protocol 3: In Vitro Skin Permeation Study
This standard protocol is used to quantitatively evaluate the permeation enhancement efficacy of IL formulations.
Materials:
- Skin Model: Excised full-thickness skin (e.g., from Yucatan micropig or rat).
- Diffusion Cells: Franz-type diffusion cells with a donor and receptor compartment.
- Test Formulation: IL-formulation containing the drug.
- Control Formulation: Aqueous or conventional formulation of the drug.
- Receptor Fluid: Buffered saline (e.g., PBS, pH 7.4), maintained at 37°C.
Procedure:
- Skin Mounting: The excised skin is carefully mounted between the donor and receptor compartments of the Franz cell, with the stratum corneum facing the donor side.
- Receptor Phase Preparation: The receptor chamber is filled with pre-warmed receptor fluid, ensuring no air bubbles are trapped under the skin.
- Formulation Application: A precise volume/weight of the test or control formulation is applied to the skin surface in the donor compartment.
- Sampling: At predetermined time intervals (e.g., 2, 4, 8, 12, 24 hours), aliquots of the receptor fluid are withdrawn and replaced with fresh fluid to maintain sink conditions.
- Analysis: The collected samples are analyzed using a validated analytical method (e.g., HPLC, UV-Vis spectroscopy) to determine the cumulative amount of drug permeated through the skin over time. Key parameters like flux (Jss) and enhancement ratio (ER) are calculated.
The Scientist's Toolkit: Essential Research Reagents
Developing and testing IL-based transdermal systems requires a specific set of reagents and materials. The following table catalogs key components and their functions in the research context.
Table 2: Key Research Reagents for IL-Based Transdermal Delivery Systems
| Reagent / Material | Function in Research & Development | Specific Examples / Notes |
|---|---|---|
| Biocompatible IL Cations | Forms the cationic moiety of the IL; chosen for low toxicity and biocompatibility [41] [42]. | Choline, 1,2-dimyristoyl-sn-glycero-3 ethyl-phosphocholine (EDMPC). |
| Biocompatible IL Anions | Forms the anionic moiety; influences solubility, stability, and permeation enhancement [41] [42]. | Amino acids (e.g., glycinate), fatty acids (e.g., linoleate), lipids. |
| Pressure-Sensitive Adhesives (PSAs) | Provides a solid matrix for patches, ensuring adhesion to skin and controlling drug release kinetics [42]. | DURO-TAK 87-4098, DURO-TAK 87-4287. Selection critical for patch stability [42]. |
| Oil Phases | Serves as the continuous medium in emulsions/dispersions; modulates drug solubility and skin partitioning [40] [42]. | Isopropyl Myristate (IPM), Octane, Hexane. |
| Surfactants & Stabilizers | Promotes the formation and stability of nanoemulsions and micelles; can influence permeation [43] [42]. | Sucrose laurate, oleylamine, oleic acid. |
| Model Biopharmaceuticals | Used as test compounds to demonstrate delivery efficacy for proteins, peptides, and nucleic acids [40] [42]. | Ovalbumin (44 kDa, model antigen), Insulin, siRNA, mRNA. |
Connecting Ionic Migration Dynamics: From PQDs to Transdermal Delivery
The core principle uniting the stability challenges in perovskite quantum dots (PQDs) and the mechanism of action of ILs in transdermal delivery is the controlled management of ionic species. In PQDs, uncontrolled migration of halide vacancies (e.g., I⁻) and A-site cations under external stimuli like electric fields or heat leads to phase segregation and material degradation [9] [39]. Strategies to suppress this involve strengthening the crystal lattice and enhancing surface ligand binding to immobilize mobile ions [39].
In a fascinating parallel, ILs are engineered to be controlled sources of mobile ions for biomedical benefit. When applied to the skin, the cationic and anionic components of the IL interact dynamically with the skin's own ionic and molecular structures. This intentional ionic migration disrupts the organized lipid lamellae of the stratum corneum, which is analogous to how mobile ions disrupt the long-range order in a perovskite lattice. The diagram below illustrates this conceptual bridge between the two fields.
Diagram: Ionic Dynamics in PQDs vs. IL-based Drug Delivery
This shared understanding of ionic dynamics provides a powerful conceptual framework. Research in PQDs focuses on immobilizing ions to preserve structure, while transdermal delivery using ILs leverages controlled ionic mobility to temporarily break down a barrier structure. Insights from one field, such as the impact of ion size and binding energy on mobility, can inform the rational design of more effective ILs in the other [9].
Ionic liquid-based platforms represent a paradigm shift in transdermal drug delivery, effectively overcoming the stratum corneum barrier for a wide range of therapeutics, from small molecules to large biopharmaceuticals. The integration of ILs into sophisticated nanocarriers and user-friendly patches has demonstrated significant success in enhancing drug stability, skin permeability, and targeted delivery for conditions like diabetes, skin cancer, and for transdermal vaccination [40] [44] [42]. The future of this field lies in the continued development of novel, highly biocompatible ILs and their integration with other physical enhancement technologies. Furthermore, the conceptual bridge to ion migration studies in materials science like perovskites promises a more profound, mechanistic understanding of how ions interact with biological barriers, paving the way for rationally designed next-generation drug delivery systems.
Solving Stability Challenges: Strategies to Control and Optimize Ion Flow
Identifying and Mitigating Primary Causes of Unwanted Ionic Migration
Ionic migration represents a fundamental challenge in the stability and performance of perovskite quantum dots (PQDs), particularly influencing their transition from laboratory research to commercial applications. This phenomenon involves the displacement of ions within the perovskite crystal structure under various external stimuli, leading to detrimental effects such as phase segregation, accelerated degradation, and inefficient charge carrier transport. Within the broader context of ionic migration dynamics in PQD surfaces research, understanding these mechanisms is paramount for developing effective mitigation strategies. The high surface-area-to-volume ratio and complex surface chemistry of PQDs render them exceptionally susceptible to ionic migration, which is often initiated or exacerbated by defects at surfaces and grain boundaries, thermal stress, and external electric fields [9].
The dynamics of ionic migration are governed by a complex interplay between intrinsic material properties and external environmental factors. At the molecular level, ionic migration in lead halide perovskites primarily involves the movement of halide anions (I-, Br-) and cation vacancies through the crystal lattice [45]. This process is facilitated by the relatively low activation energies for ion mobility within perovskite structures, making them inherently prone to ionic displacement under operational conditions. When considering PQD surfaces specifically, the presence of organic ligands and surface defects creates additional pathways for ion movement, further complicating the migration dynamics [14]. The migration not only redistributes ionic species within the material but also generates undesirable non-radiative recombination centers that capture charge carriers and reduce device efficiency [46].
Table 1: Primary Ionic Species Involved in Migration Processes in Perovskite Quantum Dots
| Ionic Species | Migration Activation Energy | Impact on Device Performance | Detection Methods |
|---|---|---|---|
| Halide Anions (I-, Br-) | Low (0.1-0.5 eV) | Phase segregation, bandgap instability, non-radiative recombination | TOF-SIMS, impedance spectroscopy [47] |
| A-site Cation Vacancies | Moderate (0.5-0.8 eV) | Hysteresis in J-V characteristics, ionic charging effects | Deep-level transient spectroscopy [46] |
| Lead Cations (Pb2+) | High (>1.0 eV) | Formation of deep-level traps, severe non-radiative losses | X-ray photoelectron spectroscopy [9] |
Fundamental Mechanisms and Driving Forces of Ionic Migration
Defect-Mediated Migration Pathways
Crystal defects serve as primary pathways for accelerated ionic migration in PQDs. Point defects, including vacancies, interstitials, and anti-site defects, create localized charge imbalances that facilitate ion movement through the crystal lattice. In mixed halide perovskites, anti-site defects such as ICs and IPb have been identified as particularly influential migration pathways [47]. These defects lower the activation energy for ion hopping between lattice sites, enabling significant ionic redistribution even at room temperature. The situation is further complicated in PQDs due to their high surface-to-volume ratio, where surface defects become increasingly dominant in the migration process. Theoretical calculations indicate that defect formation energies are significantly reduced near PQD surfaces compared to bulk regions, creating preferential pathways for ion migration along surfaces and interfaces [9].
The migration behavior varies considerably between different perovskite compositions. Research on all-inorganic mixed halide perovskites has demonstrated that specific anti-site defects create continuous pathways for halide ion migration throughout the material [47]. This defect-mediated migration follows a hop-to-hop mechanism, where ions move between adjacent defect sites under the influence of concentration gradients or external fields. The presence of these defects not only facilitates ion movement but also generates deep-level traps within the bandgap that act as non-radiative recombination centers, simultaneously degrading both stability and efficiency [46].
External Stress-Induced Migration
Beyond intrinsic defects, external stimuli significantly accelerate ionic migration in PQDs. Thermal stress, electric fields, and photoexcitation provide the necessary activation energy to overcome ionic diffusion barriers, substantially increasing migration rates. Electrically induced directional ion migration has been visualized in two-dimensional perovskite heterostructures, revealing that halide anions migrate toward the positive bias under applied electric fields [45]. This directional migration aligns with crystal and heterojunction edges, creating distinct ion migration channels that ultimately lead to compositional inhomogeneity and device performance degradation.
Thermal effects represent another critical driver of ionic migration. In situ structural analysis of CsxFA1-xPbI3 PQDs across the compositional range has demonstrated distinct thermal degradation mechanisms dependent on A-site composition [9]. Cs-rich PQDs undergo a phase transition from black γ-phase to yellow δ-phase with increasing temperature, while FA-rich PQDs with higher ligand binding energy directly decompose into PbI2. This thermal degradation is directly linked to accelerated ion migration at elevated temperatures, which is particularly problematic for PQD photovoltaic processing and operation that require elevated temperatures. Under light illumination, the situation is further exacerbated as photo-induced ion migration activates ionic vacancies and interstitial defects, leading to trap redistribution and enhanced non-radiative recombination pathways [46].
Experimental Characterization and Detection Methods
Advanced Imaging and Spectroscopy Techniques
Cutting-edge characterization methods have been developed to directly visualize and quantify ionic migration in PQD systems. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) has emerged as a powerful technique for mapping ionic distribution with high spatial resolution, enabling researchers to track halide migration pathways in mixed halide perovskites [47]. This approach has been instrumental in demonstrating how tin-lead alloying effectively suppresses ion migration by tightening the lattice structure and enhancing octahedral bonding in all-inorganic mixed halide perovskites. Complementarily, confocal microscopy imaging has revealed that halide migration channels preferentially align with crystal and heterojunction edges under external electric bias, providing visual evidence of directional ion movement [45].
In situ structural and optical characterization techniques offer real-time monitoring of ionic migration processes. Temperature-dependent X-ray diffraction (XRD) analysis across the CsxFA1-xPbI3 PQD compositional range has identified distinct thermal degradation mechanisms directly linked to ion migration [9]. Similarly, in situ photoluminescence measurements during thermal treatment have elucidated the relationship between composition, surface ligand binding, and thermal stability. For electrical characterization, galvanostatic measurements combined with optical microscopy provide quantitative assessment of ion migration rates and their impact on device performance [47]. These techniques collectively enable researchers to correlate ionic migration behavior with specific structural features and compositional elements in PQD systems.
Table 2: Experimental Methods for Characterizing Ionic Migration in PQDs
| Characterization Method | Key Measured Parameters | Spatial Resolution | Applicable Conditions |
|---|---|---|---|
| Time-of-flight secondary ion mass spectrometry (TOF-SIMS) | Ionic distribution, migration pathways | Sub-micrometer | Ex situ, depth profiling [47] |
| In situ X-ray diffraction (XRD) | Crystal structure changes, phase transitions | ~1 μm | Thermal stress, up to 500°C [9] |
| Confocal microscopy | Real-time ion migration visualization | ~200 nm | Under electric bias, illumination [45] |
| Galvanostatic measurements | Ion migration rates, activation energies | Macroscopic | Applied electric fields [47] |
| Impedance spectroscopy | Ionic conductivity, defect densities | Macroscopic | Frequency-dependent electric fields [46] |
Electrical and Thermal Analysis Methods
Electrical characterization techniques provide critical insights into the dynamics and consequences of ionic migration in operational PQD devices. The presence of mobile ions significantly influences the current-voltage (J-V) characteristics of perovskite solar cells, causing hysteresis between forward and reverse scans that directly correlates with ion migration intensity [46]. This hysteresis arises from the redistribution of ionic species under applied bias, which modifies the internal electric fields and charge collection efficiency. Detailed analysis of J-V hysteresis has revealed that reducing trap density through proper surface passivation effectively mitigates both hysteresis and trap-mediated non-radiative recombination [46].
Thermal analysis methods further illuminate the relationship between temperature and ionic migration. In situ monitoring of PQD behavior during thermal treatment has demonstrated that degradation initiation temperatures vary significantly with A-site composition and surface ligand binding energy [9]. These thermal degradation processes are directly linked to accelerated ion migration at elevated temperatures, which facilitates phase transitions in Cs-rich PQDs and direct decomposition to PbI2 in FA-rich PQDs. By combining thermal analysis with structural characterization, researchers can establish comprehensive temperature-dependent migration models that inform the development of thermally stable PQD formulations for commercial applications requiring elevated processing and operational temperatures.
Mitigation Strategies for Unwanted Ionic Migration
Compositional Engineering and Alloying Approaches
Strategic compositional engineering has proven highly effective in suppressing unwanted ionic migration in PQDs. Tin-lead (Sn-Pb) alloying for all-inorganic mixed halide perovskites demonstrates remarkable efficacy in inhibiting ion migration behavior through multiple mechanisms [47]. The incorporation of small-sized Sn²⁺ cations tightens the lattice structure, thereby enhancing Pb/Sn-X (X=I and Br) ionic bonds and effectively immobilizing halide ions. Additionally, Sn substitution significantly reduces anti-site defects such as ICs and IPb, which are considered potential pathways for ion migration. This dual approach of lattice tightening and defect reduction results in greatly suppressed ion migration, reduced hysteresis, and improved operational stability of PSC devices [47].
A-site cation engineering represents another powerful strategy for mitigating ionic migration. Research on CsxFA1-xPbI3 PQDs across the entire compositional range has revealed that FA-rich PQDs with higher ligand binding energy exhibit different thermal degradation behavior compared to Cs-rich PQDs [9]. The stronger bonding between FA cations and the perovskite lattice creates a more stable crystal structure that resists ion displacement under thermal stress. Furthermore, mixed A-site compositions can be optimized to achieve an optimal balance between phase stability and electronic properties, simultaneously addressing ionic migration concerns while maintaining high photovoltaic performance. These compositional approaches highlight the critical relationship between elemental composition, lattice dynamics, and ion migration tendencies in PQD systems.
Surface Ligand Engineering and Passivation Techniques
Surface ligand management constitutes a fundamental approach for suppressing ionic migration in PQDs. The development of an alkali-augmented antisolvent hydrolysis (AAAH) strategy has enabled significantly improved conductive capping on PQD surfaces, facilitating rapid substitution of pristine insulating oleate ligands with hydrolyzed conductive counterparts [14]. This approach creates alkaline environments that render ester hydrolysis thermodynamically spontaneous and lower reaction activation energy by approximately nine-fold, enabling the formation of dense conductive short ligand capping on the PQD surface. The resulting enhanced surface coverage effectively passivates surface defects and creates a barrier against ion migration, leading to fewer trap-states, homogeneous orientations, and minimal particle agglomerations in the assembled light-absorbing layers [14].
The strength of ligand binding to PQD surfaces plays a crucial role in determining migration resistance. First-principle density functional theory (DFT) calculations have demonstrated that the bond strength of ligands to FA-rich PQDs is larger than that for Cs-rich PQDs, illustrating the strong correlation between stability and ligand bond strength [9]. This enhanced binding creates a more robust surface layer that restricts ion movement and improves thermal tolerance. Additionally, proper ligand engineering optimizes the inter-dot spacing and electronic coupling between adjacent PQDs, facilitating efficient charge transport while simultaneously suppressing ionic migration. Through careful selection of ligand chemistry and exchange protocols, researchers can create PQD systems with significantly enhanced migration resistance without compromising electronic performance.
Interface Engineering and Processing Optimization
Interface engineering plays a crucial role in mitigating ionic migration by addressing the vulnerable boundaries between PQD layers and charge transport materials. Advanced interface engineering strategies have been developed to create barrier layers that physically block ion movement while facilitating efficient charge extraction [46]. These interfaces are specifically designed to passivate surface states and reduce trap densities, thereby eliminating the pathways for ion migration. Research has demonstrated that proper interface engineering not only suppresses ionic migration but also addresses the interconnected challenges of non-radiative recombination and J-V hysteresis, leading to simultaneous improvements in both stability and efficiency [46].
Processing optimization represents another critical dimension of migration suppression. The development of rational regulation strategies for perovskite precursor inks and deposition techniques has enabled the production of large-area perovskite films with minimized defects and enhanced crystal quality [48]. By carefully controlling parameters such as solvent selection, antisolvent rinsing protocols, and crystallization kinetics, researchers can achieve uniform, high-quality perovskite films with reduced intrinsic migration tendencies. For PQD systems specifically, the use of methyl benzoate (MeBz) with suitable polarity as an antisolvent for interlayer rinsing of PQD solid films has demonstrated superior binding and charge transfer properties on the PQD surface [14]. When combined with alkaline treatment to enhance ester hydrolysis, this processing approach enables the formation of robust conductive capping layers that effectively suppress ionic migration while maintaining excellent electronic properties.
Table 3: Comprehensive Mitigation Strategies for Ionic Migration in PQDs
| Mitigation Strategy | Key Mechanism | Experimental Implementation | Impact on Ionic Migration |
|---|---|---|---|
| Tin-Lead Alloying | Lattice tightening, enhanced ionic bonds, reduced anti-site defects | Sn²⁺ substitution in Pb-based perovskites | Significant suppression of halide migration [47] |
| A-site Cation Optimization | Improved lattice stability, enhanced ligand binding | Cs/FA/MA mixing to optimal tolerance factor | Reduced vacancy-mediated migration [9] |
| Alkali-Augmented Antisolvent Hydrolysis | Enhanced ligand exchange, dense surface capping | KOH with methyl benzoate antisolvent | ~2x ligand surface coverage, reduced surface defects [14] |
| Interface Engineering | Defect passivation, migration barrier formation | Strategic interlayer design between PQD and CTLs | Blocked interfacial migration pathways [46] |
Research Reagent Solutions for Ionic Migration Studies
Table 4: Essential Research Reagents for Ionic Migration Experiments
| Reagent/Chemical | Function in Migration Studies | Application Protocol | Key Considerations |
|---|---|---|---|
| Methyl benzoate (MeBz) | Antisolvent for interlayer rinsing, ligand source | Layer-by-layer deposition with controlled humidity | Hydrolyzes to benzoate ligands; polarity suitable for PQD integrity [14] |
| Potassium hydroxide (KOH) | Alkaline catalyst for ester hydrolysis | Coupled with ester antisolvent in AAAH strategy | Enhances hydrolysis spontaneity; lowers activation energy [14] |
| Poly-allylamine hydrochloride (PAH) | Cationic polyelectrolyte for surface charge control | Layer-by-layer deposition on membrane surfaces | Modifies surface zeta potential; affects ion depletion zones [49] |
| Polystyrene sulfonate (PSS) | Anionic polyelectrolyte for surface charge control | Alternating deposition with PAH for LbL treatment | Creates highly charged surfaces; influences ionic behavior [49] |
| Sodium dodecyl sulfate (SDS) | Ionic surfactant for SZP manipulation | Addition to solution to enhance surface charge magnitude | Forms secondary filtration layer; enhances ion depletion [49] |
The comprehensive understanding and effective mitigation of unwanted ionic migration in PQDs represent critical challenges that must be addressed to facilitate their commercial adoption in photovoltaics and other optoelectronic applications. Through detailed mechanistic studies, it has become evident that ionic migration arises from the complex interplay between intrinsic material properties (defect chemistry, lattice dynamics, surface characteristics) and external stimuli (electric fields, thermal stress, illumination). The strategies outlined in this review—including compositional engineering, surface ligand management, interface design, and processing optimization—provide a multifaceted toolkit for suppressing ionic migration and its detrimental effects on device performance and stability.
Future research directions should focus on developing even more precise control over PQD surface chemistry and interface properties to further enhance migration resistance. The integration of in situ and operando characterization techniques will provide deeper insights into the dynamic processes of ionic migration under realistic operational conditions. Additionally, the exploration of novel material systems beyond lead halide perovskites may yield fundamentally new approaches to addressing ionic migration challenges. As these research avenues progress, the field moves closer to realizing the full potential of PQD-based technologies with the stability and reliability required for widespread commercial implementation. Through continued interdisciplinary collaboration and fundamental materials research, the persistent challenge of unwanted ionic migration in PQDs can be effectively overcome, paving the way for their successful integration into the next generation of optoelectronic devices.
Surface Passivation Protocols to Heal Defects and Suppress Ion Leakage
Ionic migration dynamics at the surfaces of perovskite quantum dots (PQDs) represent a central challenge in advancing their application in optoelectronics and photovoltaics. Surface defects in these nanomaterials act as conduits for ion leakage, which accelerates non-radiative recombination, degrades photoluminescence quantum yield (PLQY), and ultimately undermines device performance and operational stability. The inherent instability arises from under-coordinated lead atoms and halide vacancies that dominate PQD surfaces, creating pathways for rapid ion diffusion. Effective surface passivation protocols are therefore critical for healing these defects and suppressing ion leakage by forming stable, protective layers that neutralize surface states and block ionic migration channels. This technical guide examines cutting-edge passivation strategies within the broader research context of controlling ionic migration dynamics in PQDs, providing researchers with experimentally-validated methodologies to enhance material performance and reliability.
Passivation Mechanisms and Defect Types
Surface defects in perovskite quantum dots primarily consist of vacancies, interstitials, and antisite defects that create charge traps and facilitate ion migration. The most prevalent defects include lead vacancies (Vₚₚ), halide vacancies (Vₓ), and under-coordinated Pb²⁺ sites, all of which act as non-radiative recombination centers and initiation points for degradation. Chemical passivation functions through Lewis acid-base interactions where electron-donating species coordinate with under-coordinated Pb²⁺ ions (Lewis acids), while field-effect passivation involves creating energy barriers that repel charge carriers from defective surfaces. A third mechanism, steric passivation, utilizes bulky organic molecules to physically block moisture and oxygen ingress while suppressing ionic diffusion through spatial constraints.
The interplay between these mechanisms and ionic migration dynamics is crucial; unpassivated surfaces exhibit accelerated halide ion migration under operational stressors, leading to phase segregation and accelerated degradation. Effective passivation protocols must address both the electronic defects (responsible for recombination losses) and ionic defects (responsible for leakage and degradation) simultaneously to achieve optimal performance.
Experimental Protocols for Surface Passivation
Sulfur-Based Passivation for Semiconductor QDs
This protocol details a two-step passivation process optimized for near-surface semiconductor quantum dots, demonstrating significant improvements in resonance fluorescence properties by reducing surface state density and electric field fluctuations [50].
Materials and Equipment:
- (NH₄)₂S aqueous solution (20%)
- Atomic layer deposition (ALD) system
- Glove box with inert atmosphere (H₂O and O₂ < 1 ppm)
- 0.02-μm syringe filters
- Al₂O₃ ALD precursor
- Sample substrate with QDs
Procedure:
- Place the glove box under inert atmosphere and maintain H₂O and O₂ levels below 1 ppm throughout the process.
- Filter the (NH₄)₂S aqueous solution using a 0.02-μm syringe filter to remove polysulfide particles.
- Immerse the sample in 20% (NH₄)₂S solution for 10 minutes at room temperature.
- Transfer the sample directly to the load-lock chamber of the ALD system without breaking the inert atmosphere.
- Deposit 10 nm of Al₂O₃ at 150°C using ALD to encapsulate the passivated surface.
- Characterize passivation effectiveness through X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy to confirm reduced surface state density.
Key Considerations: The critical innovation lies in maintaining an unbroken inert atmosphere between the sulfur treatment and ALD encapsulation, preventing reoxidation and ensuring uniform passivation layer formation.
Alkaline-Augmented Antisolvent Hydrolysis for PQDs
This protocol enhances conductive capping on PQD surfaces through alkaline-facilitated ligand exchange, substantially improving charge transfer and suppressing ion leakage [14].
Materials and Equipment:
- Methyl benzoate (MeBz) antisolvent
- Potassium hydroxide (KOH)
- PQD colloids (e.g., FA₀.₄₇Cs₀.₅₃PbI₃)
- Spin coater
- Centrifuge
Procedure:
- Prepare hybrid FA₀.₄₇Cs₀.₅₃PbI₃ PQDs with average size of ~12.5 nm via post-synthetic cation exchange.
- Create an alkaline environment by adding KOH to methyl benzoate antisolvent.
- Spin-coat PQD colloids into solid films using standard parameters.
- Rinse the PQD solid films with the KOH-modified methyl benzoate antisolvent.
- The alkaline environment facilitates rapid hydrolysis of the ester, generating conductive ligands that replace pristine insulating oleate ligands.
- Complete the process with a post-treatment using short cationic ligands (e.g., FA⁺) to substitute pristine oleylammonium ligands on the A-site.
Key Considerations: The alkaline environment reduces the activation energy for ester hydrolysis by approximately 9-fold, enabling nearly double the conventional amount of conductive ligands to cap the PQD surface, which significantly reduces trap states and minimizes particle agglomeration.
Dual-Interface Passivation with Piperazinium Salts
This protocol addresses defective interfaces in wide-bandgap perovskite films through a top-down dual-interface carrier management strategy, effectively suppressing ion migration at both top and buried interfaces [51].
Materials and Equipment:
- Piperazinium chloride
- Halide-substituted piperazinium salts (Cl⁻, Br⁻, I⁻)
- Wide-bandgap perovskite precursor solution (1.68 eV)
- ITO substrates
- Spin coater
Procedure:
- Synthesize halide ion-substituted piperazinium salts as post-treatment modifiers.
- Prepare wide-bandgap perovskite films (1.68 eV) on appropriate substrates.
- Treat the perovskite surface with piperazinium chloride solution via spin coating.
- The large piperazinium cations concentrate at the perovskite top surface, suppressing nonradiative recombination and improving energy band alignment.
- Simultaneously, the small chloride anions migrate downward to accumulate at the buried interface.
- This creates asymmetric bidirectional ion distribution, achieving dual-interface defect passivation.
Key Considerations: The differential migration of cationic and anionic components enables comprehensive interface healing, with the chloride anions forming strong Pb-Cl bonds at the buried interface that effectively suppress iodine migration and vacancy formation.
Quantitative Analysis of Passivation Efficacy
Table 1: Performance Metrics of Passivated Quantum Dot Systems
| Passivation Method | Material System | Key Performance Improvement | Stability Enhancement | Reference |
|---|---|---|---|---|
| Sulfur-based + ALD | InAs/GaAs QDs | RF linewidth reduction: 43.23±22.53 to 19.68±6.48 GHz | Revived pulsed-RF signals in previously non-emitting QDs | [50] |
| Alkaline-augmented antisolvent | FA₀.₄₇Cs₀.₅₃PbI₃ PQDs | Certified PCE: 18.3% (record for PQDSCs) | Improved storage and operational stability | [14] |
| Piperazinium chloride dual-interface | 1.68 eV WBG perovskite | PCE: 22.3%, VOC × FF product: 84.4% of SQ limit | 91.3% initial efficiency after 1200 h MPPT | [51] |
| Core-shell thickness optimization | NaGdF₄:Yb,Er@NaGdF₄ | Upconversion enhancement: 290-fold; Downshifting: 25-fold | Linear lifetime increase with shell thickness | [52] |
Table 2: Defect Reduction Metrics Across Passivation Strategies
| Passivation Strategy | Defect Type Addressed | Ion Leakage Suppression Mechanism | Characterization Techniques |
|---|---|---|---|
| Sulfur-based chemical passivation | Surface dangling bonds | Reduced surface state density and electric field | XPS, Raman spectroscopy, RF linewidth measurement [50] |
| Conductive ligand exchange | Halide vacancies, lead vacancies | Hydrolyzed conductive ligands seal surface vacancies | FTIR, TOF-SIMS, DEMS [14] |
| Dual-interface engineering | Interface defects, cation vacancies | Bidirectional ion distribution heals both interfaces | In-situ DEMS, DFT calculations [51] |
| Core-shell architecture | Surface quenching centers | Spatial isolation of core from environment | TEM, HAADF-STEM, time-resolved spectroscopy [52] |
Visualization of Passivation Mechanisms
Sulfur Passivation Process Flow
Diagram 1: Sulfur-based passivation workflow emphasizing critical inert atmosphere transfer to prevent reoxidation before final encapsulation [50].
Alkaline-Augmented Antisolvent Mechanism
Diagram 2: Alkaline-augmented antisolvent hydrolysis mechanism showing enhanced ligand exchange for superior surface capping and ion leakage suppression [14].
Dual-Interface Passivation Dynamics
Diagram 3: Dual-interface passivation dynamics showing differential migration of piperazinium cations and chloride anions for comprehensive interface healing [51].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Surface Passivation Research
| Reagent | Function | Application Context | Key Properties |
|---|---|---|---|
| (NH₄)₂S (20% aqueous) | Sulfur-based passivator | Semiconductor QD surface defect termination | Eliminates surface dangling bonds, reduces state density [50] |
| Methyl benzoate with KOH | Alkaline-augmented antisolvent | PQD interlayer rinsing | Facilitates ester hydrolysis, conductive ligand exchange [14] |
| Piperazinium chloride | Dual-interface modifier | Wide-bandgap perovskite surface treatment | Asymmetric ion distribution, top-bottom passivation [51] |
| NaGdF₄ shell precursors | Core-shell passivation | Lanthanide-doped nanoparticle surfaces | Spatial isolation, quenching suppression [52] |
| 9-Anthracenecarboxylic acid | Molecular sensitizer | LnNP-molecule hybrid systems | Triplet energy transfer, optimized at 0.8 nm shell thickness [52] |
Surface passivation protocols have emerged as indispensable tools for healing defects and suppressing ion leakage in perovskite quantum dots and related nanomaterials. The methodologies detailed herein—from sulfur-based chemical passivation to alkaline-augmented ligand exchange and dual-interface engineering—provide robust experimental frameworks for addressing the fundamental challenge of ionic migration dynamics in PQD systems. The quantitative improvements in device performance and stability metrics underscore the critical importance of targeted surface engineering in advancing quantum dot technologies toward commercial viability.
Future research directions should prioritize the development of multifunctional passivators that simultaneously address multiple defect types while providing energy level alignment, the establishment of standardized accelerated aging protocols to predict long-term stability under operational conditions, and the creation of computational screening methods to identify novel passivation molecules with optimal binding energies and steric properties. As the field progresses, integrating these advanced passivation strategies with scalable fabrication techniques will be essential for translating laboratory successes into commercially viable quantum dot technologies with predictable performance and durability.
Compositional Engineering (A-Site, X-Site) for Intrinsically Stable Surfaces
The dynamic nature of ionic migration across perovskite quantum dot (PQD) surfaces represents a fundamental challenge to the operational stability and commercial viability of perovskite-based optoelectronics. This technical review examines how compositional engineering at the A-site and X-site of the perovskite lattice serves as a foundational strategy to suppress ionic migration and cultivate intrinsically stable surfaces. By systematically modulating cation and anion constituents, researchers can directly influence the activation energy for defect formation and ion mobility, thereby enhancing the thermodynamic and electrochemical resilience of PQDs. This review synthesizes recent advances in lead-free compositions, mixed-cation/halide formulations, and surface ligand manipulations, providing a structured analysis of quantitative stability metrics and detailed experimental methodologies for evaluating ionic migration dynamics.
Metal halide perovskites (MHPs) possess an inherently ionic crystal structure (ABX₃), which facilitates remarkable optoelectronic properties but also predisposes them to ion migration under operational stressors such as electric fields, light, and heat [53]. In perovskite quantum dots (PQDs), this phenomenon is exacerbated by the immense surface-to-volume ratio, where under-coordinated surface ions act as primary pathways for uncontrolled migration. The photoinduced migration of halide ions, in particular, leads to phase segregation in mixed-halide perovskites, forming iodide-rich and bromide-rich domains that act as charge carrier traps and degrade device performance [53]. Furthermore, the thermodynamic and redox characteristics of halide perovskites create a strong driving force for hole trapping and the oxidation of iodide species, making the mobility of halide ions and their vulnerability to hole-induced oxidation major factors governing long-term stability [53].
Compositional engineering directly targets the root causes of ionic mobility. By tailoring the elemental constituents at the A-site (e.g., Cs⁺, MA⁺, FA⁺) and X-site (e.g., I⁻, Br⁻, Cl⁻, pseudo-halides), it is possible to strengthen the perovskite lattice, enhance defect formation energies, and passivate surface vacancies that serve as initiation points for degradation. This approach works in concert with surface ligand engineering to create a synergistic effect, where a stable internal lattice is protected by a robust, passivating surface layer [54]. The following sections delve into the specific strategies for A-site and X-site engineering, supported by quantitative data and experimental protocols.
A-Site Cation Engineering for Enhanced Lattice Stability
The A-site cation, while not directly contributing to the frontier electronic orbitals, plays a critical role in stabilizing the perovskite lattice by influencing the tilting of the [BX₆]⁴⁻ octahedra and determining the tolerance factor, a key metric for structural stability.
Monovalent Cation Tuning and Mixed-Cation Strategies
Pure FAPbI₃ possesses a near-ideal tolerance factor but is thermodynamically unstable at room temperature, prone to transitioning from the black perovskite phase (α-phase) to a non-perovskite yellow phase (δ-phase). Conversely, CsPbI₃ suffers from even greater phase instability despite its all-inorganic nature. Research on CsₓFA₁₋ₓPbI₃ PQDs across the entire compositional range has revealed that the thermal degradation mechanism is intrinsically linked to the A-site composition.
- Cs-Rich PQDs (e.g., CsPbI₃): Thermal degradation is initiated by a phase transition from the black γ-phase to the yellow δ-phase [55].
- FA-Rich PQDs (e.g., FAPbI₃): Exhibit superior thermal stability against phase transitions and directly decompose into PbI₂ at higher temperatures without passing through a yellow phase intermediary [55].
This enhanced stability in FA-rich PQDs is attributed to stronger ligand binding energy. Density functional theory (DFT) calculations confirm that the bond strength of common ligands (e.g., oleylamine, oleic acid) to the surface of FA-rich PQDs is larger than that for Cs-rich PQDs, thereby providing a more effective kinetic barrier against surface erosion and ion migration [55]. Furthermore, FA-rich PQDs exhibit stronger electron–longitudinal optical (LO) phonon coupling, which influences charge carrier dynamics [55].
Table 1: Quantitative Impact of A-Site Composition on Stability and Optoelectronic Properties of CsₓFA₁₋ₓPbI₃ PQDs
| Composition | Thermal Degradation Pathway | Ligand Binding Energy | Electron-LO Phonon Coupling | Phase Stability |
|---|---|---|---|---|
| Cs-Rich (e.g., CsPbI₃) | Phase transition (γ- to δ-phase) then decomposition [55] | Weaker [55] | Weaker [55] | Lower (prone to δ-phase) [55] |
| FA-Rich (e.g., FAPbI₃) | Direct decomposition to PbI₂ [55] | Stronger [55] | Stronger [55] | Higher (stable black phase) [55] |
Lead-Free A-Site Compositions: Bismuth as a Case Study
The drive to eliminate toxic lead has led to the exploration of alternative B-site cations, with bismuth (Bi³⁺) emerging as a promising candidate. Bismuth-based PQDs, such as Cs₃Bi₂Br₉, offer a fundamentally different chemical profile.
- Intrinsic Stability: Lead-free Cs₃Bi₂Br₉ PQDs demonstrate extended serum stability and already meet current safety standards without additional coating, overcoming the lead-related toxicity barriers of CsPbBr₃ PQDs [13].
- Performance: Despite typically lower power conversion efficiencies (PCEs) compared to lead-based counterparts, bismuth-based photoelectrochemical sensors have demonstrated exceptional sensitivity, achieving sub-femtomolar detection of microRNA (miRNA) [13].
X-Site Anion Engineering to Suppress Halide Migration
The X-site anion is a primary participant in the ionic migration process. Engineering at this site focuses on filling halide vacancies and increasing the activation energy for halide ion movement.
Halide Alloying and Pseudo-Halide Incorporation
Mixed-halide perovskites (e.g., CsPbIₓBr₃₋ₓ) are essential for bandgap tuning, but they are susceptible to photoinduced phase segregation. A powerful strategy to suppress this is the incorporation of pseudo-halide anions, which are polyatomic anions that mimic halides but often form stronger bonds with the perovskite lattice.
A notable example is the use of the fluorinated pseudo-halide anion hexafluorophosphate (PF₆⁻). Its introduction as a surface ligand for FAPbI₃ PQDs via a post-synthetic ligand exchange strategy has yielded remarkable results [56]:
- Multi-Functional Passivation: PF₆⁻ simultaneously passivates iodide vacancies, minimizes inter-dot spacing for enhanced electronic coupling, suppresses ion migration, and provides a hydrophobic barrier [56].
- Performance and Stability: This synergistic effect enabled FAPbI₃ PQD solar cells with an unprecedented PCE of 19.01% and significantly enhanced storage and operational stability [56].
Table 2: Impact of X-Site Engineering with Pseudo-Halide Anions on PQD Properties
| Pseudo-Halide Anion | Ionic Radius | Primary Functions | Demonstrated Outcome |
|---|---|---|---|
| Hexafluorophosphate (PF₆⁻) | 2.38 Å [56] | Passivates iodide vacancies, enhances electronic coupling, suppresses ion migration, provides hydrophobicity [56] | 19.01% PCE in FAPbI₃ PQD solar cells; enhanced operational stability [56] |
Experimental Protocols for Investigating Compositional Stability
Validating the efficacy of compositional engineering strategies requires a suite of advanced characterization techniques to probe ionic migration and surface stability directly.
In Situ Spectroscopic and Structural Characterization
Protocol: In Situ XRD for Thermal Degradation Analysis
- Objective: To monitor phase and structural changes in PQDs in real-time under thermal stress.
- Methodology:
- PQD films are deposited on a thermally stable substrate (e.g., Pt) inside an X-ray diffractometer equipped with a heating stage.
- The temperature is ramped from room temperature (e.g., 30 °C) to a high temperature (e.g., 500 °C) under an inert atmosphere (e.g., argon flow) to prevent oxidation.
- XRD patterns are continuously collected at set temperature intervals.
- Data Analysis: The appearance, shift, or disappearance of diffraction peaks is tracked. For example, the decomposition of FAPbI₃ PQDs is identified by the emergence of PbI₂ peaks at ~25.2°, 29.0°, and 41.2° without a prior phase transition, while Cs-rich PQDs show a shift from γ-phase to δ-phase peaks before decomposition [55].
Protocol: In Situ Photoluminescence (PL) under Electrical Bias
- Objective: To assess phase segregation in mixed-halide PQDs induced by electric fields and ion migration.
- Methodology:
- A mixed-halide PQD (e.g., CsPbIₓBr₃₋ₓ) film is integrated into a simple device structure with transparent electrodes.
- The film is placed under a spectrometer and an external electric bias is applied.
- PL spectra are collected continuously during biasing.
- Data Analysis: The onset and evolution of new, red-shifted PL peaks indicate the formation of iodide-rich domains due to halide segregation. The kinetics of this process and its reversibility serve as a direct metric for the efficacy of compositional engineering in suppressing ion migration [53].
Theoretical Modeling of Surface Ligand Interactions
Protocol: DFT Calculations for Ligand Binding Energy
- Objective: To quantitatively compare the strength of ligand binding to different PQD surfaces at the atomic level.
- Methodology:
- Model surfaces of different A-site compositions (e.g., CsPbI₃ vs. FAPbI₃) are constructed, including common surface terminations and defects.
- Ligand molecules (e.g., oleylamine, oleic acid, PF₆⁻) are positioned at their most probable binding sites.
- The binding energy (Eb) is calculated as Eb = E(PQD+ligand) - EPQD - E_ligand, where E denotes the total energy of each system from DFT simulations.
- Data Analysis: A more negative E_b indicates a stronger and more stable bond. This computational approach directly explained the superior thermal stability of FA-rich PQDs, as calculations showed a larger ligand binding energy compared to Cs-rich surfaces [55].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and materials essential for implementing the compositional engineering strategies discussed in this review.
Table 3: Essential Research Reagents for PQD Compositional Engineering and Stability Research
| Reagent / Material | Function in Research | Technical Note |
|---|---|---|
| Cesium Precursors (e.g., Cs₂CO₃, Cs-oleate) | A-site precursor for all-inorganic CsPbX₃ PQDs [54] [17] | Enables synthesis of Cs-rich PQDs; critical for studying phase stability of γ-CsPbI₃. |
| Formamidinium Precursors (e.g., FAI, FAPbI₃) | A-site precursor for hybrid organic-inorganic FAPbI₃ PQDs [55] [56] | Essential for creating FA-rich compositions with enhanced thermal and phase stability. |
| Bismuth Salts (e.g., BiBr₃, BiI₃) | B-site precursor for lead-free PQDs (e.g., Cs₃Bi₂Br₉) [13] | Offers a non-toxic alternative; provides intrinsic stability and meets safety standards. |
| Pseudo-Halide Salts (e.g., HPF₆, NH₄PF₆) | Source of PF₆⁻ for surface ligand exchange and vacancy passivation [56] | Multifunctional anion for suppressing iodide vacancies and ion migration. |
| Long-Chain Ligands (e.g., Oleic Acid, Oleylamine) | Capping ligands for colloidal synthesis and surface stabilization [54] [17] | Dynamic binding nature requires engineering; foundational for nanoparticle dispersion and initial growth. |
| Polar Solvents (e.g., Hexane, Ethyl Acetate) | Purification and ligand exchange solvents [54] | Inevitably impact PQD structural integrity; choice and exposure time must be carefully optimized. |
Visualization of Experimental Workflows and Relationships
The following diagrams illustrate the core experimental workflow for surface engineering and the logical relationship between compositional choices and stability outcomes.
Workflow for Stable PQD Surfaces
Compositional Stability Strategy Map
Compositional engineering at the A-site and X-site is not merely a tool for bandgap tuning but a profound intervention in the ionic dynamics of perovskite quantum dots. By moving beyond simple CsPbX₃ formulations toward sophisticated mixed-cation (e.g., CsₓFA₁₋ₓPbI₃) and pseudo-halide-integrated (e.g., PF₆⁻) structures, researchers can directly target the thermodynamic and kinetic drivers of ionic migration. The synergistic combination of a stabilized bulk lattice through A-site tuning and a passivated, vacancy-free surface through X-site engineering presents the most promising path toward achieving intrinsically stable PQD surfaces. This approach, validated by advanced in situ characterization and theoretical modeling, is critical for advancing PQD technologies from laboratory curiosities toward robust commercial applications in photovoltaics, light-emitting diodes, and biosensing. Future research must continue to bridge the gap between atomic-scale understanding of ion dynamics and macroscopic device performance and lifetime.
Ligand Exchange and Conductive Capping for Robust and Stable PQD Surfaces
The performance and operational stability of perovskite quantum dot (PQD) optoelectronic devices are predominantly governed by the intricate chemistry of their surfaces. While the bulk perovskite core often exhibits exceptional optoelectronic properties, the high surface-to-volume ratio of quantum dots means that surface states, capped with long-chain insulating ligands, can severely impede charge carrier transport between adjacent dots. Furthermore, these surface ligand layers are dynamic, and their instability is a primary source of defect formation and uncontrolled ionic migration. Ionic migration, particularly of halide anions, is a critical degradation pathway that leads to phase segregation, increased non-radiative recombination, and eventual device failure [45]. This technical guide examines advanced ligand exchange and conductive capping strategies as direct mechanisms to suppress ionic migration by passivating surface vacancies and creating robust, thermodynamically stable PQD surfaces. The subsequent sections detail the latest experimental protocols, quantitatively compare their outcomes, and provide a toolkit for researchers to implement these surface engineering techniques, all within the critical context of stabilizing PQDs against ionic migration.
Experimental Protocols for Advanced Ligand Exchange
This section provides detailed methodologies for two cutting-edge ligand exchange strategies that have demonstrated high efficacy in producing efficient and stable PQD solar cells.
Consecutive Surface Matrix Engineering (CSME) for FAPbI₃ PQDs
The CSME protocol is designed to disrupt the dynamic equilibrium of native ligands and occupy resulting surface vacancies with short-chain conjugated ligands [57].
- Materials: FAPbI₃ PQDs stabilized with Oleic Acid (OA) and Oleylamine (OAm); short-chain conjugated ligands (e.g., thiophene-based molecules like 2-thiophenemethylammonium iodide (2-TM) or 2-thiopheneethylammonium iodide (2-TE)).
- Procedure:
- Solution-Phase Ligand Exchange: The FAPbI₃ PQD solution is treated with the short-chain conjugated ligand. The mixture is stirred at 60-80°C for 30-60 minutes to initiate the replacement of long-chain OA ligands.
- Induction of Amidation: The reaction environment is controlled to induce an amidation reaction between the displaced OA and OAm. This reaction pulls the equilibrium, favoring the desorption of these insulating ligands from the PQD surface.
- Purification: The PQDs are purified by adding an antisolvent (e.g., methyl acetate) followed by centrifugation to remove the displaced ligand byproducts.
- Solid-State Ligand Exchange (Optional): The spin-cast PQD solid film can be further treated by a post-deposition rinse with a solution of the short-chain ligand to ensure complete surface coverage and defect passivation [58].
Alkali-Augmented Antisolvent Hydrolysis (AAAH) for Interlayer Rinsing
The AAAH strategy enhances the conventional ester-based rinsing process by creating an alkaline environment that drastically improves the hydrolysis efficiency, leading to a denser conductive capping layer [14] [59].
- Materials: Methyl benzoate (MeBz) antisolvent; Potassium Hydroxide (KOH); Layer-by-layer deposited PQD solid films (e.g., FA₀.₄₇Cs₀.₅₃PbI₃).
- Procedure:
- Preparation of Alkaline Antisolvent: A small, optimized concentration of KOH (e.g., 0.05-0.1 mM) is dissolved in methyl benzoate antisolvent. The alkaline environment is critical to render ester hydrolysis thermodynamically spontaneous and lower the reaction activation energy.
- Layer-by-Layer Rinsing: Each layer of the spin-cast PQD solid film is rinsed with the KOH/MeBz solution during the layer-by-layer deposition process.
- Hydrolysis and Ligand Exchange: Upon exposure to ambient humidity, the alkaline antisolvent rapidly hydrolyzes the methyl benzoate into benzoate anions. These short-chain conductive anions efficiently substitute the pristine insulating oleate (OA⁻) ligands on the PQD surface.
- Evaporation: The antisolvent is swiftly removed by spin-coating and evaporation, leaving behind a densely packed PQD film with a high coverage of conductive benzoate capping ligands.
Quantitative Performance Comparison of Ligand Engineering Strategies
The following table summarizes the performance outcomes of recent advanced ligand exchange strategies, providing a clear benchmark for researchers.
Table 1: Performance Metrics of PQD Solar Cells via Advanced Ligand Engineering
| Ligand Strategy | PQD Material | Key Function | Best PCE (%) | Stability Performance | Ref. |
|---|---|---|---|---|---|
| Consecutive Surface Matrix Engineering (CSME) | FAPbI₃ | Disrupts OA/OAm equilibrium, occupies vacancies | 19.14 | Improved operational stability | [57] |
| Alkali-Augmented Antisolvent Hydrolysis (AAAH) | FA₀.₄₇Cs₀.₅₃PbI₃ | Enhances ester hydrolysis for dense conductive capping | 18.37 (Certified 18.30) | Improved storage & operational stability | [14] [59] |
| Fluorinated Pseudo-Halide Anion (PF₆⁻) | FAPbI₃ | Passivates vacancies, enhances coupling, provides hydrophobicity | 19.01 | Enhanced storage & operational stability | [56] |
| Dual-Phase Synergistic Ligand Exchange (DSLE) | FAPbI₃ | Replaces insulating ligands & passivates defects | 18.21 | >80% initial efficiency after 1400 h in ambient | [58] |
The Impact of Ligand Engineering on Ionic Migration Pathways
The primary objective of robust conductive capping is to suppress ionic migration, a key failure mechanism in PQD devices. The diagram below illustrates how effective ligand engineering directly influences these migration pathways.
Figure 1: A comparison of ionic migration dynamics in PQD films with poor versus engineered surface capping. Effective ligand exchange directly passivates surface vacancies, which are the primary channels for ion migration [45] [56].
As illustrated in Figure 1, ineffective ligand capping leaves behind a high density of surface vacancies. These vacancies act as channels for halide anions (e.g., I⁻) to migrate under the influence of external electric fields, light, or heat [45]. This migration leads to deleterious effects, including phase segregation, increased trap-assisted non-radiative recombination, and significant device degradation. Conversely, strategies like CSME and the use of fluorinated pseudo-halide anions (PF₆⁻) efficiently occupy these surface vacancies with strongly-bound ligands [57] [56]. This robust capping creates a physical and energetic barrier that suppresses the initiation and propagation of ionic migration, thereby enhancing the device's operational stability.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their functions for implementing the discussed ligand exchange and surface passivation protocols.
Table 2: Essential Reagents for PQD Surface Ligand Engineering
| Reagent | Function in Experiment | Key Property / Rationale |
|---|---|---|
| Methyl Benzoate (MeBz) | Alkaline-augmented antisolvent for interlayer rinsing. | Moderate polarity preserves PQD core; hydrolyzes into conductive benzoate ligands [14]. |
| Potassium Hydroxide (KOH) | Additive to ester antisolvents to create an alkaline environment. | Facilitates spontaneous ester hydrolysis; lowers activation energy for ligand exchange [14] [59]. |
| Short-Chain Conjugated Ligands (e.g., 2-TM, 2-TE) | Dual-site molecular ligands for solution and solid-phase exchange. | Replace insulating OA/OAm; passivate defects via coordination with Pb²⁺; enhance electronic coupling [58]. |
| Fluorinated Pseudo-Halide (e.g., PF₆⁻) | Multifunctional anionic surface ligand. | Large ionic radius minimizes inter-dot spacing; passivates iodide vacancies; provides hydrophobicity [56]. |
| Trioctylphosphine Oxide (TOPO) | Surface passivating ligand for CsPbI₃ PQDs. | Coordinates with undercoordinated Pb²⁺ ions to suppress non-radiative recombination [60]. |
| Oleic Acid (OA) / Oleylamine (OAm) | Native, insulating ligands from synthesis. | Dynamically bound; require replacement or modification to enable charge transport [57] [14]. |
Optimizing Environmental and Operational Parameters to Minimize Degradation
In the broader context of ionic migration dynamics across perovskite quantum dot (PQD) surfaces, understanding and mitigating degradation is paramount for advancing their application in optoelectronics and photovoltaics. The ionic nature of PQDs renders them susceptible to degradation under environmental stressors such as humidity, oxygen, heat, and light, as well as operational stressors like external electric bias [13] [61]. This degradation is intrinsically linked to ion migration, which can lead to phase segregation, defect formation, and eventual performance failure [45]. This technical guide synthesizes current research to provide a comprehensive framework of optimization strategies, detailing experimental protocols and material solutions to suppress ion migration and enhance the operational lifetime of PQD-based devices.
Key Degradation Mechanisms and the Role of Ion Migration
The degradation of PQDs is primarily driven by their low formation energy and ionic crystal structure, which facilitate the migration of halide anions and A-site cations under various stimuli [61] [45].
- Electrically Induced Ion Migration: The application of an external electric field can induce directional migration of halide ions (e.g., I⁻, Br⁻) within the perovskite lattice. Studies on two-dimensional perovskite heterostructures have visualized halide anions migrating toward the positive bias, with migration channels often aligning with crystal edges and heterojunctions, leading to compositional inhomogeneity and device asymmetry [45].
- Environmental Degradation: Exposure to moisture and oxygen can accelerate ion migration and lead to the hydrolysis of the perovskite crystal structure. Furthermore, the intrinsic dynamic binding of surface ligands can result in their desorption, creating unprotected surface sites that are vulnerable to attack and facilitating further ion leakage [13] [14].
- Thermal and Photo-Induced Degradation: Elevated temperatures and prolonged illumination provide the activation energy required for ions to overcome migration barriers, leading to defect formation and phase transitions that quench photoluminescence and degrade electronic performance [60] [61].
Optimizing Environmental Parameters
Advanced Encapsulation and Surface Passivation
Creating a physical barrier against environmental stressors is a critical strategy. Atomic Layer Deposition (ALD) has emerged as a superior technique for depositing conformal, pinhole-free protective layers.
Experimental Protocol: Low-Temperature ALD of Alumina on CsPbBr₃ QDs [62]
- Substrate Preparation: Spin-coat a film of CsPbBr₃ PQDs onto a clean glass substrate.
- ALD Chamber Setup: Place the substrate in the ALD chamber. Use Trimethylaluminum (TMA) and H₂O as precursors, with nitrogen as the carrier gas.
- Temperature Optimization: Set the deposition temperature to 75°C. This temperature was identified as optimal, balancing film quality with the thermal sensitivity of PQDs. Temperatures at or above 80°C were found to cause significant lattice damage.
- Deposition Cycle: Execute multiple ALD cycles (e.g., 50-100 cycles). Each cycle consists of:
- A pulsed exposure to TMA.
- A purging step to remove excess precursor.
- A pulsed exposure to H₂O.
- A final purging step.
- Post-Deposition Analysis: Characterize the encapsulated PQDs using photoluminescence (PL) spectroscopy and X-ray diffraction (XRD) to confirm enhanced stability without compromised optical properties.
Table 1: Impact of ALD Temperature on CsPbBr₃ PQD Properties
| ALD Temperature (°C) | Photoluminescence (PL) Intensity | Environmental Stability | Structural Integrity |
|---|---|---|---|
| 40 - 70 | Moderate to High Retention | Good Improvement | Preserved |
| 75 | High Retention | Excellent Improvement | Fully Preserved |
| ≥ 80 | Significant Reduction | Poor Improvement | Severe Damage |
Ligand Engineering for Intrinsic Stability
Modifying the surface ligands of PQDs is a chemical approach to passivate surface defects and suppress ion migration by providing a stable, conductive capping layer.
Experimental Protocol: Alkaline-Augmented Antisolvent Hydrolysis (AAAH) [14]
- PQD Film Deposition: Spin-coat a solid film of hybrid FA₀.₄₇Cs₀.₅₃PbI₃ PQDs.
- Antisolvent Preparation: Prepare a methyl benzoate (MeBz) antisolvent solution containing a controlled concentration of potassium hydroxide (KOH). The alkaline environment facilitates the rapid hydrolysis of the ester into benzoate ligands.
- Interlayer Rinsing: During the layer-by-layer film assembly, rinse the freshly deposited PQD solid film with the KOH/MeBz antisolvent. This step substitutes the pristine, long-chain insulating oleate (OA⁻) ligands with the in-situ hydrolyzed short-chain benzoate ligands.
- A-site Ligand Exchange: As a subsequent step, a post-treatment with a solution of short cationic ligands (e.g., formamidinium iodide) in 2-pentanol can be applied to exchange the original A-site cations, further enhancing inter-dot charge transport [14].
This AAAH strategy has been shown to double the amount of conductive ligands on the PQD surface, resulting in films with fewer trap states, minimal agglomeration, and significantly improved photovoltaic performance and operational stability [14].
Optimizing Operational Parameters
Compositional Engineering to Suppress Ion Migration
Multicomponent perovskites, where the A-, B-, and X-sites are occupied by a mixture of ions, have proven effective in increasing the activation energy for ion migration, thereby stabilizing the lattice [61].
Table 2: Multicomponent Perovskite Compositions for Enhanced Stability
| Perovskite Site | Example Components | Function in Stability Enhancement |
|---|---|---|
| A-Site | Cs⁺, MA⁺, FA⁺, Rb⁺ | Synergistic size compensation adjusts the Goldschmidt tolerance factor to stabilize the photoactive α-phase at room temperature [61]. |
| B-Site | Pb²⁺, Sn²⁺ | Mixing metals can tune the bandgap, but stability often remains a challenge [61]. |
| X-Site | I⁻, Br⁻, Cl⁻ | Partial substitution of I⁻ with smaller Br⁻ or Cl⁻ can compensate for A-site changes and directly suppress halide anion migration by increasing migration activation energy [61]. |
An exemplary stable composition is Cs₀.₀₅(FA₀.₈₃MA₀.₁₇)₀.₉₅Pb(I₀.₈₃Br₀.₁₇)₃, which leverages multi-site mixing for optimal stability [61].
Managing Electrical and Thermal Stress
Operational stability requires careful management of applied fields and temperature.
- Electric Field Management: The observation of electrically induced directional ion migration underscores the need for device operational protocols that minimize constant high bias. Pulsed electrical operation, rather than continuous DC bias, could help mitigate cumulative ion displacement and the resulting device degradation [45].
- Thermal Management: Operational temperatures should be maintained well below the phase transition thresholds of the specific PQD composition. For example, CsPbI₃ PQDs are susceptible to a structural phase transition at temperatures around 180°C, leading to severe PL quenching [60]. Effective heat dissipation in device architectures is non-negotiable for long-term operation.
Experimental Methodologies for Characterization
A robust experimental workflow is essential for systematically investigating PQD degradation and the efficacy of optimization strategies.
Key Characterization Techniques:
- In-situ/Ion Migration Visualization: As demonstrated in 2D perovskite heterostructures, confocal microscopy can be used to visualize real-time halide anion migration under electrical bias, identifying preferential migration channels along crystal edges [45].
- Electrochemical Impedance Spectroscopy (EIS): EIS is a powerful tool for probing ion transport dynamics within functional devices. Analysis of the Péclet number from EIS data can reveal the coexistence of diffusive and convective ion transport regimes, informing better device design [63].
- Photoluminescence (PL) Tracking: Monitoring the PL intensity, quantum yield (PLQY), and emission wavelength under continuous illumination or thermal stress provides a direct measure of optical stability and defect formation [60] [62].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for PQD Stability Research
| Reagent | Function/Application | Key Detail |
|---|---|---|
| Methyl Benzoate (MeBz) | Antisolvent for interlayer rinsing | Hydrolyzes into conductive benzoate ligands for X-site capping; moderate polarity preserves PQD core [14]. |
| Trioctylphosphine Oxide (TOPO) | Surface ligand for defect passivation | Coordinates with undercoordinated Pb²⁺ ions; shown to enhance PL intensity by 18% in CsPbI₃ PQDs [60]. |
| Potassium Hydroxide (KOH) | Alkaline catalyst for AAAH strategy | Facilitates spontaneous ester hydrolysis in antisolvent, enabling efficient ligand exchange [14]. |
| Trimethylaluminum (TMA) | Precursor for ALD encapsulation | Reacts with H₂O to form a conformal Al₂O₃ barrier layer at optimized low temperatures (~75°C) [62]. |
| Formamidinium Iodide (FAI) | A-site cation source for post-treatment | Substitutes pristine OAm⁺ ligands to improve electronic coupling between PQDs [14]. |
| Lead Bromide (PbBr₂) | Precursor for inorganic framework | Key component in synthesizing CsPbBr₃ PQDs; high purity (99.99%) is crucial for low defect density [62]. |
The path to minimizing degradation in perovskite quantum dots is multifaceted, requiring an integrated approach that addresses both environmental and operational parameters. The strategies outlined—ranging from low-temperature ALD encapsulation and advanced ligand engineering to multicomponent compositional design—are unified by their common goal of suppressing ionic migration dynamics. The experimental protocols and characterization methodologies provided herein offer a reproducible framework for researchers to diagnose degradation pathways and validate new stabilization techniques. As the field progresses, the synergy between advanced material synthesis, precise encapsulation technologies, and sophisticated operando characterization will be instrumental in unlocking the full commercial potential of PQD technologies.
Benchmarking Performance: Validating Stability and Functional Efficacy
Ionic migration is a critical phenomenon influencing the performance and stability of perovskite quantum dots (PQDs). The inherent ionic nature of lead halide perovskite crystals, characterized by their ABX3 structure (where A is a monovalent cation such as cesium or formamidinium, B is lead, and X is a halide anion), facilitates the mobility of ions under external biases or during operational stress [14] [15]. This ionic movement primarily occurs through defect sites, such as vacancies and interstitials, and along grain boundaries and PQD surfaces where the crystal lattice is discontinuous. On PQD surfaces, the large surface-to-volume ratio intensifies these effects, as the surface strain and incomplete ligand capping create numerous pathways for ion migration [14].
Understanding and quantifying this migration is paramount for advancing PQD applications in photovoltaics, light-emitting diodes, and quantum information technologies. Uncontrolled ion migration leads to non-radiative recombination, phase segregation, and efficiency losses in devices like PQD solar cells (PQDSCs) [14] [15]. This technical guide, framed within the broader thesis of ionic migration dynamics in PQD surfaces research, provides a comprehensive framework for measuring the fundamental quantitative metrics of this process: activation energy and ionic conductivity. By establishing standardized experimental protocols and data interpretation methods, this guide aims to enable researchers to accurately characterize and ultimately mitigate the detrimental effects of ion migration, paving the way for more stable and efficient PQD-based devices.
Core Quantitative Metrics and Theoretical Framework
The movement of ions within a solid material like a perovskite is a thermally activated process. Its kinetics and scale are fundamentally described by two interlinked quantitative metrics: the activation energy for migration and the ionic conductivity.
Activation Energy (Ea)
The activation energy for ion migration (Ea) represents the minimum energy barrier an ion must overcome to move from one stable lattice site to another. It is a definitive measure of the mobility of ionic species within the crystal. A lower Ea indicates easier ion migration, which is often associated with material instability, while a higher Ea suggests a more stable lattice with restricted ion movement. The activation energy is intrinsically linked to the strength of chemical bonds and the geometry of the migration pathway. It can be experimentally determined from the temperature dependence of ionic conductivity or directly from current transients, as detailed in Section 3.
Ionic Conductivity (σ)
The ionic conductivity (σ) quantifies a material's ability to conduct electric current via the movement of ions rather than electrons. It is a bulk property that depends on both the number of mobile ions and their mobility. Ionic conductivity is not a fixed value but follows the Arrhenius law, exhibiting a strong, predictable dependence on temperature:
[ \sigma = \frac{\sigma0}{T} \exp\left(-\frac{Ea}{k_B T}\right) ]
where (\sigma0) is the pre-exponential factor, (T) is the absolute temperature, (kB) is the Boltzmann constant, and (E_a) is the activation energy. This relationship is the cornerstone for extracting Ea from conductivity measurements.
Table 1: Key Quantitative Metrics for Ion Migration Analysis
| Metric | Symbol | Definition | Typical Units | Significance in PQDs |
|---|---|---|---|---|
| Activation Energy | (E_a) | Energy barrier for ion hopping | eV (electronvolts) | Determines ion mobility under operational stress; lower (E_a) implies higher instability. |
| Ionic Conductivity | (\sigma) | Measure of current carried by ions | S/cm (Siemens per centimeter) | Indicates the density of mobile ions and their ease of movement. |
| Pre-exponential Factor | (\sigma_0) | Material-specific constant related to charge carrier density | K S/cm | Related to the number of attempting ions; can reveal conduction mechanism. |
The interaction of these metrics defines the ionic landscape of a material. For instance, strategies that enhance the conductive capping on PQD surfaces, such as the alkaline treatment substituting insulating oleate ligands with shorter, hydrolyzed counterparts, have been shown to reduce trap-states and minimize particle agglomeration [14]. This surface modification likely passivates ionic migration pathways, which would be reflected in an increased measured activation energy and reduced ionic conductivity.
Experimental Protocols for Measurement
Accurate measurement requires carefully designed experiments and controlled environments to isolate ionic current from electronic contributions.
Direct Current (DC) Polarization Method
This method is ideal for materials where ionic conductivity is significantly higher than electronic conductivity.
- Objective: To measure the steady-state ionic current and calculate bulk ionic conductivity.
- Procedure:
- Device Fabrication: Prepare a metal-insulator-metal (MIM) structure. For PQDs, this involves depositing a uniform, thick film (e.g., ~1 µm) of PQDs between two inert, blocking electrodes (e.g., Au, Pt) to prevent electrode reactions from influencing the measurement.
- Measurement: Apply a constant DC bias voltage (e.g., 0.1 - 0.5 V) across the device and measure the current as a function of time using a source-meter unit.
- Data Analysis: The current will decay over time as mobile ions migrate and are blocked at the electrodes, leading to ionic polarization. The steady-state current ((I{ss})) is dominated by electronic leakage. The ionic conductivity ((\sigma{ionic})) is calculated using the formula derived from Ohm's law: [ \sigma{ionic} = \frac{I{ss} \cdot L}{V \cdot A} ] where (L) is the film thickness, (V) is the applied voltage, and (A) is the electrode area.
Thermal Activation Energy Measurement
This protocol builds upon conductivity measurement to extract the activation energy.
- Objective: To determine the activation energy ((E_a)) for ion migration from the temperature dependence of ionic conductivity.
- Procedure:
- Temperature Control: Place the sample in a temperature-controlled stage or probe station, capable of operating over a defined range (e.g., 300 K to 350 K).
- Ionic Conductivity Profiling: At each stabilized temperature point, perform the DC polarization measurement (as in 3.1) to determine the ionic conductivity ((\sigma_{ionic})).
- Arrhenius Plotting: Plot (ln(\sigma \cdot T)) versus (1/T). The data should conform to a straight line according to the Arrhenius equation.
- Activation Energy Calculation: Perform a linear fit to the data. The activation energy ((Ea)) is derived from the slope of the fitted line: [ \text{Slope} = -\frac{Ea}{kB} ] where (kB) is the Boltzmann constant (8.617333262145 × 10⁻⁵ eV/K).
Table 2: Summary of Key Experimental Methodologies
| Method | Primary Output | Key Advantages | Key Limitations |
|---|---|---|---|
| DC Polarization | Bulk Ionic Conductivity ((\sigma)) | Simple setup; directly measures ionic current. | Requires ion-blocking electrodes; less accurate if electronic conductivity is high. |
| Thermal Activation | Activation Energy ((E_a)) | Provides fundamental insight into migration energy barriers. | Requires precise temperature control; assumes a single activation energy dominates. |
Diagram 1: Workflow for measuring Ea and conductivity.
The Scientist's Toolkit: Essential Research Reagents and Materials
The quality of PQD samples and the choice of characterization tools are critical for obtaining reliable data on ion migration.
Table 3: Key Research Reagent Solutions for PQD Ion Migration Studies
| Category | Specific Example | Function in Research |
|---|---|---|
| PQD Precursors | Cesium lead halides (e.g., CsPbI₃, CsPbBr₃), Formamidinium lead halides (FA₀.₄₇Cs₀.₅₃PbI₃) [14] | Forms the core perovskite crystal structure under investigation for ion migration dynamics. |
| Ligands for Surface Capping | Oleic acid/Oleate (OA), Potassium hydroxide (KOH) for alkaline treatment [14], Methyl benzoate (MeBz) antisolvent [14] | Passivates surface defects and controls the surface chemistry. Conductive ligands replace insulating ones to reduce trap states and potentially alter ion migration pathways. |
| Antisolvents | Methyl acetate (MeOAc), Methyl benzoate (MeBz) [14] | Used in layer-by-layer deposition to remove pristine ligands and induce ligand exchange during film formation, affecting film morphology and defect density. |
| Electrode Materials | Gold (Au), Platinum (Pt) | Serve as inert, blocking electrodes in MIM capacitor structures for electrical characterization of ionic transport. |
| Additives for Defect Passivation | Uracil [15], Guanabenz acetate salt [15] | Strengthen grain boundaries and passivate ionic defects (vacancies), thereby directly suppressing the sources of ion migration. |
Data Interpretation and Advanced Analysis
Interpreting the raw data from electrical measurements requires understanding the underlying physical processes. The Arrhenius plot ((ln(σT)) vs. (1/T)) is the primary tool. A single, straight line suggests one dominant ion migration mechanism. However, deviations from linearity or the presence of two distinct slopes can indicate multiple migration mechanisms with different activation energies—for instance, bulk ion migration versus grain boundary-assisted migration. In PQDs, where surfaces and interfaces dominate, grain boundary and surface migration often have a lower Ea than lattice migration.
Correlating electrical metrics with material properties is crucial. For example, a study on hybrid FA₀.₄₇Cs₀.₅₃ PQDs using an alkali-augmented antisolvent hydrolysis (AAAH) strategy reported fewer trap-states and more homogeneous films [14]. One would expect such a sample to exhibit a higher measured activation energy (Ea) for ion migration compared to a control sample with inefficient ligand exchange, as the conductive capping more effectively passivates surface vacancies that serve as ion migration pathways.
Furthermore, the pre-exponential factor (\sigma0) can provide insights. An unusually high or low (\sigma0) may indicate a change in the number of charge carriers or the attempt frequency for ion jumps, often linked to changes in defect chemistry induced by different synthesis or treatment methods.
Diagram 2: Data interpretation pathways for ion migration.
Comparative Analysis of Different PQD Compositions and Surface Treatments
Perovskite quantum dots (PQDs) represent a transformative class of materials with exceptional optoelectronic properties, including tunable bandgaps, high photoluminescence quantum yields, and defect tolerance. Their performance in applications ranging from photovoltaics to biosensing is critically dependent on two interrelated factors: core composition and surface treatment. This whitepaper provides a systematic analysis of lead-based and lead-free PQD variants, examining how different surface engineering strategies—including ligand exchange, passivation, and alkaline treatments—mitigate instability and ionic migration issues. By synthesizing recent research advances, we demonstrate that rational material design can simultaneously enhance device performance, operational stability, and environmental safety, paving the way for next-generation PQD technologies.
Perovskite quantum dots have emerged as promising materials for advanced optoelectronic applications due to their exceptional properties such as high absorption coefficients, tunable bandgaps, and high charge carrier mobility. The general formula of these materials is ABX₃, where A is an organic cation (e.g., MA⁺, FA⁺) or inorganic cation (e.g., Cs⁺), B is a metal cation (typically Pb²⁺ or Sn²⁺), and X is a halide anion (I⁻, Br⁻, Cl⁻). The compositional versatility of PQDs enables precise tuning of their electronic and optical properties, making them suitable for applications including photovoltaics, light-emitting diodes, and biosensors [13] [14].
A critical challenge in PQD technology is the inherent instability of the perovskite crystal structure, particularly under environmental stressors such as moisture, oxygen, and electric fields. Ionic migration within the crystal lattice, especially halide anion mobility, leads to phase segregation, degradation, and performance hysteresis in devices [45]. This ionic drift-diffusion dynamics represents a fundamental limitation for practical implementations. Surface chemistry plays a pivotal role in modulating these dynamics, as the PQD surface serves as both a gateway for environmental degradation and a platform for engineering enhanced stability [14].
This review systematically analyzes the relationship between PQD core composition, surface treatment methodologies, and the resulting material properties, with particular emphasis on controlling ionic migration for improved device performance and longevity.
Comparative Analysis of PQD Compositions
The electronic structure, stability, and toxicity profiles of PQDs are fundamentally determined by their elemental composition. Research has primarily focused on lead-halide perovskites, though increasing environmental concerns have accelerated the development of lead-free alternatives.
Table 1: Comparison of Key PQD Compositions and Their Properties
| Composition | Bandgap (eV) | Photoluminescence Quantum Yield (%) | Stability | Toxicity Concerns | Primary Applications |
|---|---|---|---|---|---|
| CsPbBr₃ | ~2.3 | Up to 85 [64] | Moderate; degrades in water | High lead content | Photovoltaics [14], Biosensing [64] |
| FA₀.₄₇Cs₀.₅₃PbI₃ | ~1.5-1.6 | High (hybrid A-site) [14] | Moderate; improved phase stability | High lead content | Photovoltaics (certified 18.3% efficiency) [14] |
| Cs₃Bi₂Br₉ (Bismuth-based) | Wider than Pb counterparts | Lower than Pb-based PQDs | High; extended serum stability [13] | Low; Bi is non-toxic [13] | Biosensing (sub-femtomolar miRNA detection) [13] |
| Lead-free Cs₃Bi₂Br₉ | Not specified | Not specified | High aqueous stability [13] | Negligible [13] | Photoelectrochemical sensors [13] |
Lead-Based Perovskite Compositions
Lead-based PQDs, particularly cesium lead halide (CsPbX₃) and formamidinium-cesium lead halide (FA-CsPbX₃) variants, currently deliver the highest performance metrics in optoelectronic applications. CsPbBr₃ PQDs exhibit outstanding photoluminescence quantum yields (PLQY) of up to 85% with narrow emission spectra, making them ideal for light-emitting applications [64]. Hybrid A-site compositions like FA₀.₄₇Cs₀.₅₃PbI₃ combine the thermal stability of Cs⁺ with the phase stability of FA⁺, enabling certified solar cell efficiencies of 18.3% [14].
The primary limitation of lead-based PQDs is their susceptibility to environmental degradation, particularly in aqueous environments. Additionally, lead toxicity presents significant concerns for commercial applications, especially in biomedical and consumer technologies. Studies indicate that Pb²⁺ release from CsPbBr₃ PQDs typically exceeds permitted levels for parenteral administration, creating regulatory barriers [13].
Lead-Free Perovskite Compositions
Lead-free alternatives aim to address the toxicity issues of conventional PQDs while maintaining reasonable performance characteristics. Among these, bismuth-based compositions such as Cs₃Bi₂Br₉ have emerged as promising candidates. These materials offer significantly enhanced stability under physiological conditions, with demonstrated longevity in serum environments [13]. Critically, bismuth-based PQDs already meet current safety standards without requiring additional coating procedures, presenting a substantial advantage for biomedical applications such as biosensing [13].
The trade-off for this improved safety profile is generally lower optoelectronic performance compared to lead-based counterparts. Bismuth-based PQDs typically exhibit wider bandgaps and lower photoluminescence quantum yields, limiting their efficiency in photovoltaic applications. However, their sufficient performance in photoelectrochemical sensing applications, achieving sub-femtomolar sensitivity for miRNA detection, demonstrates their practical utility in specialized domains [13].
Surface Treatment Methodologies
Surface treatments represent critical strategies for enhancing PQD stability and modulating ionic migration without altering core composition. These methodologies primarily focus on ligand engineering and interface modification to passivate surface defects and strengthen the PQD structure against environmental degradation.
Table 2: Surface Treatment Methods for PQDs
| Treatment Method | Mechanism of Action | Impact on Stability | Effect on Performance | Limitations |
|---|---|---|---|---|
| Alkali-Augmented Antisolvent Hydrolysis (AAAH) | Facilitates ester hydrolysis for ligand exchange; increases conductive capping [14] | Improces operational stability [14] | Certified 18.3% solar cell efficiency [14] | Requires precise control of alkalinity |
| Conventional Ester Antisolvent Rinsing | Substitutes pristine OA- ligands with hydrolyzed counterparts [14] | Moderate improvement | Limited by weak ligand binding [14] | Inefficient hydrolysis; extensive surface defects [14] |
| Conductive COF Integration | Provides stable, ordered scaffold for PQDs; enables π-π stacking [64] | Enhanced stability over 30 days [64] | Ultrasensitive dopamine detection (0.3 fM) [64] | Complex synthesis process |
| Surface Passivation | Reduces surface defects and ion migration pathways [13] | Extends stability for weeks [13] | Improved charge transfer | Does not fully address lead leaching |
Ligand Exchange and Engineering
Ligand exchange processes are fundamental to PQD surface functionalization, replacing native insulating ligands with shorter conductive alternatives to enhance inter-dot charge transfer. Conventional approaches utilizing ester antisolvents like methyl acetate (MeOAc) rely on ambient hydrolysis to generate short-chain acetate ligands that substitute the pristine oleate (OA⁻) capping layer [14]. However, this method suffers from inefficient hydrolysis under ambient conditions, resulting in incomplete ligand exchange and extensive surface vacancy defects that capture charge carriers and facilitate ionic migration [14].
The recently developed Alkali-Augmented Antisolvent Hydrolysis (AAAH) strategy represents a significant advancement by creating alkaline environments that fundamentally alter the thermodynamics and kinetics of ester hydrolysis. Theoretical calculations reveal that alkaline conditions render ester hydrolysis thermodynamically spontaneous and lower the reaction activation energy by approximately 9-fold [14]. Using potassium hydroxide (KOH) coupled with methyl benzoate (MeBz) antisolvent, this approach enables rapid substitution of insulating oleate ligands with up to twice the conventional amount of hydrolyzed conductive counterparts [14]. The resulting PQD films exhibit fewer trap states, homogeneous orientations, and minimal particle agglomerations, leading to certified solar cell efficiencies of 18.3% - the highest among hybrid A-site PQD solar cells [14].
Matrix Encapsulation and Integration
Encapsulating PQDs within stabilizing matrices provides physical protection against environmental degradation while maintaining electronic coupling. Covalent Organic Frameworks (COFs) have emerged as particularly effective host matrices due to their highly ordered porous architectures and π-conjugated systems. The integration of CsPbBr₃ PQDs within a COF matrix creates a synergistic nanocomposite that leverages the optical strengths of PQDs and the structural advantages of COFs [64].
This approach enables the fabrication of dual-mode sensing platforms that combine fluorescence quenching and electrochemical impedance spectroscopy (EIS) for ultrasensitive dopamine detection with limits of detection reaching 0.3 fM (fluorescence) and 2.5 fM (EIS) [64]. The COF matrix protects PQDs from aggregation and degradation in aqueous environments while facilitating selective molecular interactions through π-π stacking and hydrogen bonding. The resulting nanocomposites maintain excellent stability over 30 days with minimal performance degradation, demonstrating the efficacy of matrix encapsulation for enhancing PQD longevity [64].
Experimental Protocols for Key Methodologies
Synthesis of CsPbBr₃ PQDs via Hot-Injection Method
Materials: Lead(II) bromide (PbBr₂, 99.999%), cesium bromide (CsBr, 99.9%, anhydrous), oleic acid (OA, technical grade, 90%), oleylamine (OAm, 80-90%), N,N-dimethylformamide (DMF, anhydrous, 99.8%), toluene (anhydrous, 99.8%) [64].
Procedure:
- Dissolve 0.085 g of CsBr (0.4 mmol) and 0.147 g of PbBr₂ (0.4 mmol) in 10 mL of anhydrous DMF under vigorous magnetic stirring in a three-neck flask equipped with a condenser and nitrogen inlet.
- Degas the mixture with high-purity nitrogen at room temperature for 15 minutes to eliminate residual oxygen and water vapor.
- Inject 1 mL of oleic acid (OA) and 0.5 mL of oleylamine (OAm) as capping ligands to stabilize the crystal surface.
- Gradually heat the mixture to 120°C (ramp rate: 5°C min⁻¹) under continuous nitrogen flow.
- Rapidly inject 0.5 mL of preheated toluene (60°C) using a syringe pump to trigger instantaneous nucleation of CsPbBr₃ nanocrystals.
- Allow the reaction to proceed for exactly 10 seconds before quenching in an ice-water bath to preserve size uniformity.
- Purify the resulting colloidal dispersion by centrifugation at 10,000 rpm for 5 minutes, followed by two washes with anhydrous toluene to remove unbound ligands.
- Redisperse the final product in 5 mL of anhydrous DMF for characterization and further use [64].
Characterization: The as-prepared PQDs should exhibit intense green emission under UV light (365 nm), a photoluminescence quantum yield of approximately 85%, and a sharp emission peak centered at 515 nm, indicating quantum confinement and high crystallinity [64].
Alkali-Augmented Antisolvent Hydrolysis (AAAH) Treatment
Materials: Methyl benzoate (MeBz), potassium hydroxide (KOH), hybrid FA₀.₄₇Cs₀.₅₃PbI₃ PQDs, anhydrous solvents.
Procedure:
- Synthesize FA₀.₄₇Cs₀.₅₃PbI₃ PQDs with an average size of ~12.5 nm via post-synthetic cation exchange of CsPbI₃ PQD parent according to established protocols [14].
- Prepare an alkaline antisolvent solution by adding controlled amounts of KOH to methyl benzoate. The alkalinity must be carefully regulated to ensure adequate ligand exchange without compromising the structural integrity of the PQDs.
- Spin-coat the resulting PQD colloids into solid films for subsequent analysis.
- Rinse the PQD solid films with the KOH-modified methyl benzoate antisolvent under ambient conditions (relative humidity ~30%).
- Control the rinsing parameters to ensure uniform coverage and rapid evaporation, facilitating the hydrolysis of ester groups and subsequent ligand exchange.
- Characterize the treated films using FTIR to confirm ligand exchange and X-ray diffraction to verify structural integrity [14].
Characterization: Successful implementation yields PQD films with enhanced conductive capping, evidenced by reduced trap states, homogeneous crystallographic orientations, and minimal particle agglomerations. Photovoltaic devices fabricated using this method should achieve efficiencies approaching 18.37% (certified 18.30%) [14].
CsPbBr₃-PQD-COF Nanocomposite Fabrication
Materials: 1,3,5-tris(4-aminophenyl)benzene (TAPB, 97%), 2,5-dihydroxyterephthalaldehyde (DHTA, 95%), CsPbBr₃ PQDs, dimethylformamide (DMF), glacial acetic acid, rhodamine B.
Procedure:
- Synthesize the covalent organic framework (COF) via Schiff-base condensation by dissolving 0.035 g of TAPB (0.1 mmol) and 0.025 g of DHTA (0.15 mmol) in 5 mL anhydrous DMF.
- Add 100 μL of glacial acetic acid as a catalyst and stir the reaction mixture at ambient temperature for 2 hours, forming a bright yellow suspension indicative of extended π-conjugation and framework formation.
- Confirm successful COF formation through characterization of characteristic C=N and C-O bands in FTIR and a sharp (100) peak at 2θ ≈ 5.8° in XRD.
- Integrate pre-synthesized CsPbBr₃ PQDs into the COF matrix by combining the materials in solution under controlled conditions.
- Incorporate rhodamine B as a visual indicator for qualitative monitoring of dopamine-induced color changes.
- Deposit the resulting CsPbBr₃-PQD-COF nanocomposite onto electrode surfaces for sensor fabrication [64].
Characterization: The nanocomposite should exhibit a green-to-pink color shift under ambient light conditions at dopamine concentrations above 100 pM, providing visual confirmation of analyte detection in addition to quantitative fluorescence and electrochemical measurements [64].
Visualization of Surface Treatment Workflows
The workflow illustrates the sequential surface engineering process for enhancing PQD performance. Beginning with as-synthesized PQDs containing native insulating ligands (OA⁻/OAm⁺), the first critical step involves rinsing with alkaline antisolvent (KOH + MeBz), which facilitates efficient hydrolysis and initiates ligand exchange [14]. This is followed by the formation of a conductive capping layer through substitution with short-chain ligands, dramatically improving inter-dot charge transfer. Subsequent integration within a COF matrix provides structural stability and additional functionalization capabilities [64]. The final result is a stable PQD film with enhanced optoelectronic properties, reduced ionic migration, and improved environmental resistance.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for PQD Surface Engineering Research
| Reagent/Category | Function | Example Applications | Key Considerations |
|---|---|---|---|
| Methyl Benzoate (MeBz) | Antisolvent for interlayer rinsing; precursor for conductive ligands via hydrolysis [14] | Alkali-augmented antisolvent hydrolysis (AAAH) [14] | Moderate polarity preserves PQD structure while enabling ligand exchange |
| Potassium Hydroxide (KOH) | Alkaline additive to enhance ester hydrolysis kinetics and thermodynamics [14] | Creating alkaline environments for efficient ligand exchange [14] | Concentration must be controlled to avoid PQD degradation |
| Covalent Organic Frameworks (COFs) | Porous, ordered matrices for PQD encapsulation and stabilization [64] | Dual-mode sensing platforms; enhanced environmental stability [64] | π-conjugated systems enable charge transfer and analyte interactions |
| Oleic Acid / Oleylamine | Native capping ligands for PQD synthesis and stabilization [64] | Initial synthesis of colloidal PQDs; surface passivation [64] | Dynamic binding enables exchange but creates instability |
| Polyethyleneimine (PEI) | Adhesion-enhancing interlayer for electrode interfaces [65] | Improving interfacial adhesion in electrode configurations | Positively charged groups interact with polar surfaces [65] |
| Rhodamine B | Visual indicator for colorimetric sensing readouts [64] | Dopamine sensing platforms providing visual concentration cues [64] | Enables green-to-pink shift at specific analyte thresholds |
The comparative analysis of PQD compositions and surface treatments reveals a complex interplay between material intrinsic properties and engineered interfaces. Lead-based PQDs currently deliver superior optoelectronic performance, with CsPbBr₃ and hybrid A-site variants achieving remarkable efficiencies in photovoltaic and sensing applications. However, their susceptibility to environmental degradation and significant toxicity concerns necessitate robust surface engineering strategies. The development of lead-free alternatives, particularly bismuth-based compositions, offers promising pathways toward environmentally benign PQD technologies with enhanced stability in biological environments.
Surface treatment methodologies, especially advanced ligand exchange techniques like alkali-augmented antisolvent hydrolysis and matrix encapsulation using COFs, have demonstrated remarkable effectiveness in mitigating ionic migration and enhancing PQD stability. These approaches address fundamental challenges in PQD technology by passivating surface defects, strengthening interfacial adhesion, and providing physical protection against environmental stressors.
Future research directions should focus on scalable lead-free formulations that bridge the performance gap with lead-based counterparts, while further refining surface treatment protocols for enhanced reproducibility and commercial viability. The integration of machine learning-assisted characterization, as demonstrated in multiplexed bacterial detection systems [13], presents promising opportunities for accelerated material optimization. Standardized validation protocols and increased attention to regulatory requirements will be essential for translating PQD technologies from laboratory demonstrations to practical applications in sensing, energy, and biomedical devices.
Correlating Suppressed Ionic Migration with Enhanced Device Performance and Lifetime
Ionic migration in perovskite quantum dots (PQDs) represents a fundamental challenge that impedes the commercial viability of next-generation optoelectronic devices. This whitepaper synthesizes current research demonstrating that suppressed ionic migration directly correlates with enhanced device performance metrics and operational longevity. Through advanced surface modification techniques, optimized ligand engineering, and strategic interface design, researchers have achieved remarkable improvements in photoluminescence quantum yield, device efficiency, and environmental stability. The findings compiled herein establish a clear structure-property-performance relationship, providing a robust framework for the rational design of high-performance, durable PQD-based devices.
Metal halide perovskites (MHPs) have emerged as revolutionary materials in optoelectronics due to their exceptional properties, including long carrier lifetimes, high defect tolerance, tunable bandgaps, and high absorption coefficients [66] [67]. Despite rapid advancements in power conversion efficiency (PCE), which now reaches certified values of 25.5% in perovskite solar cells (PSCs), operational instability remains a critical barrier to commercialization [66]. This instability largely originates from the inherent ionic nature of perovskites, which exhibit mixed ionic-electronic conductivity under device-operating conditions [1] [67].
The soft ionic lattice of halide perovskites enables significant ion migration when subjected to external stimuli such as electric fields, light, heat, and environmental factors [1]. Under operating conditions, ions present in the perovskite bulk migrate toward electrodes due to electrostatic effects, reacting with interfacial layers and triggering device degradation pathways [66]. This ionic movement leads to numerous detrimental phenomena including current-voltage hysteresis, phase segregation in mixed-halide perovskites, accelerated interfacial degradation, and ultimately device failure [66] [67]. While early research focused primarily on extrinsic stability issues (e.g., protection from moisture and oxygen), recent investigations have revealed that intrinsic instability from ion migration persists even in encapsulated devices, presenting a more fundamental challenge [66].
This whitepaper examines the critical correlation between suppressed ionic migration and enhanced device performance and lifetime within the broader context of ionic migration dynamics in PQD surfaces research. By synthesizing findings from recent studies encompassing material synthesis, advanced characterization, and device engineering, we establish a comprehensive framework linking specific suppression strategies to quantifiable improvements in key performance metrics.
Ionic Migration Fundamentals in Perovskite Systems
Mechanisms and Primary Migrating Species
Ionic migration in perovskite structures occurs primarily through vacancy-mediated diffusion mechanisms, where ions hop between lattice sites via point defects [68] [67]. Theoretical studies utilizing density functional theory (DFT) have revealed that different ionic species exhibit markedly different migration activation energies, leading to varied migration rates under operational conditions [68].
Experimental evidence from Rutherford backscattering spectroscopy (RBS) depth profiling of aged inorganic perovskite solar cells (CsPbI₂Br) has directly identified iodide ions (I⁻) as the most mobile species, followed by cesium ions (Cs⁺) [66]. This non-destructive technique revealed significant concentration gradients of I⁻ across the perovskite layer thickness in aged devices, indicating substantial migration even without external stress. Complementary simulation studies of CH₃NH₃PbI₃ (MAPbI₃) perovskite solar cells have further identified iodine and methylammonium (MA⁺) vacancies as the fastest migrating defects due to their low migration barriers (approximately 0.1–0.6 eV) [68].
Table 1: Primary Migrating Species in Metal Halide Perovskites
| Ionic Species | Migration Barrier (eV) | Relative Migration Rate | Experimental Evidence |
|---|---|---|---|
| Iodide ions (I⁻) | 0.1-0.6 [68] | Highest | RBS depth profiling [66] |
| Cs⁺ ions | Moderate | Medium | RBS depth profiling [66] |
| MA⁺ vacancies | 0.1-0.6 [68] | High | Computational simulation [68] |
| Pb²⁺ ions | High | Lowest | RBS showing negligible mobility [66] |
Impact on Device Performance and Degradation
Ionic migration influences device performance through multiple mechanisms that operate across different timescales. In the short term, mobile ions accumulate at interfaces and grain boundaries, forming charge-blocking barriers that impede carrier transport and cause current-voltage hysteresis [68] [66]. This phenomenon is particularly pronounced in hybrid organic-inorganic perovskites containing volatile organic ions like methylammonium (MA⁺) when exposed to ambient air, humidity, or heat [66].
Over extended operational periods, ionic migration leads to irreversible device degradation through several pathways. The interfacial diffusion of halide ions into adjacent charge transport layers creates defects and non-radiative recombination centers, reducing both open-circuit voltage and fill factor [66]. Simulation studies have demonstrated that ion accumulation near interfaces can reach concentrations approaching 10¹⁸ cm⁻³ under stress conditions (500°C and 1V bias), severely degrading carrier transport and accelerating device failure [68]. Additionally, in mixed-halide systems, photoinduced ion segregation leads to phase separation, altering local bandgaps and reducing photocurrent generation efficiency [69].
Quantitative Correlation: Suppression Strategies and Device Outcomes
Recent research has established definitive correlations between specific ion migration suppression strategies and enhanced device performance metrics. The data compiled in Table 2 demonstrates consistent improvements across multiple material systems and device architectures.
Table 2: Correlation Between Ionic Migration Suppression Strategies and Device Performance Enhancements
| Suppression Strategy | Material System | Ionic Migration Reduction | Performance Improvement | Lifetime Enhancement |
|---|---|---|---|---|
| TOPO surface passivation [60] | CsPbI₃ PQDs | Non-radiative recombination suppression | PLQY increase: 18% [60] | - |
| PF₆⁻ ligand engineering [56] | FAPbI₃ PQDs | Iodide vacancy passivation, suppressed ion migration | PCE: 19.01% (vs. uncontrolled) [56] | Enhanced operational stability [56] |
| RGO electrode implementation [68] | CH₃NH₃PbI₃ PSCs | Interfacial ion concentration reduced by order of magnitude | Improved stability under bias | Reduced degradation rate [68] |
| FA-rich composition [69] | FA₀.₆MA₀.₄PbI₃ | Iodide migration from bulk to surface passivates vacancies | Longer carrier lifetime vs. MA-rich [69] | Improved surface stability |
| L-PHE modification [60] | CsPbI₃ PQDs | Surface defect passivation | PL intensity retention: >70% after 20 days UV [60] | Superior photostability |
Surface Ligand Engineering
Surface ligand manipulation has emerged as a particularly effective strategy for suppressing ionic migration in PQDs. A systematic investigation of CsPbI₃ PQDs demonstrated that surface passivation using trioctylphosphine oxide (TOPO), trioctylphosphine (TOP), and l-phenylalanine (L-PHE) effectively suppressed non-radiative recombination through coordination with undercoordinated Pb²⁺ ions and surface defects [60]. The corresponding photoluminescence (PL) enhancements of 18%, 16%, and 3% were observed for TOPO, TOP, and L-PHE, respectively, directly correlating with improved surface defect passivation [60]. Notably, L-PHE-modified PQDs demonstrated exceptional photostability, retaining over 70% of their initial PL intensity after 20 days of continuous UV exposure [60].
The strategic replacement of traditional oleate ligands with multifunctional fluorinated pseudo-halide anions such as hexafluorophosphate (PF₆⁻) in FAPbI₃ PQDs has yielded remarkable device performance improvements [56]. Leveraging its coordination capability, large ionic radius (2.38 Å), and intrinsic hydrophobicity, PF₆⁻ simultaneously passivates iodide vacancies, minimizes inter-dot spacing for enhanced electronic coupling, suppresses ion migration, and provides a hydrophobic barrier [56]. This comprehensive approach achieved an unprecedented PCE of 19.01% for FAPbI₃ PQD solar cells, with enhanced storage and operational stability [56].
Compositional Engineering
Cation selection and stoichiometry play crucial roles in modulating ionic migration dynamics. Comparative studies of mixed-cation single crystals (FA₀.₆MA₀.₄PbI₃ vs. FA₀.₄MA₀.₆PbI₃) have revealed that higher formamidinium (FA) content significantly influences both defect formation and ion migration behavior [69]. Density functional theory (DFT) calculations demonstrate that samples with higher FA content have a lower energy barrier for iodide ions to migrate from the bulk to the top layer, assisting in passivating surface vacancies, and a higher energy diffusion barrier to escape from surface to vacuum [69]. This dual effect results in fewer surface vacancies and longer-lived hole-electron pairs, explaining the superior performance of FA-rich compositions [69].
The replacement of volatile organic cations (MA⁺) with inorganic counterparts (Cs⁺) has also shown significant promise in reducing ionic migration. Inorganic halide perovskites (IHPs) such as CsPbI₂Br generally exhibit improved operational stability and reduced current-voltage hysteresis compared to their hybrid counterparts [66]. However, RBS studies confirm that ion migration still occurs in aged IHP devices, with I⁻ and Cs⁺ ions showing detectable mobility even without external stress [66]. This highlights that while compositional engineering improves stability, supplemental suppression strategies remain necessary.
Interface and Electrode Engineering
The choice of electrode materials and interface design significantly influences ionic migration patterns and device degradation pathways. Comparative simulations of CH₃NH₃PbI₃ perovskite solar cells with silver versus reduced graphene oxide (RGO) back contacts revealed striking differences in ion accumulation behavior [68]. Devices incorporating RGO electrodes demonstrated interfacial ion concentrations lowered by an order of magnitude compared to conventional silver contacts, promoting more uniform ionic distributions across the device [68]. This reduction in interfacial ion accumulation directly correlated with improved operational stability and reduced degradation rates.
The mechanism behind this improvement involves the suppression of ionic build-up near interfaces and grain boundaries, which typically impedes carrier transport and accelerates cell degradation [68]. Under stress conditions (500°C and 1V bias), simulations showed that ion accumulation peaks at approximately 10¹⁸ cm⁻³ within 1 hour in devices with silver electrodes, while RGO contacts substantially mitigated this effect [68]. This research highlights the critical importance of electrode selection in managing ionic migration for enhanced device lifetime.
Experimental Protocols for Ionic Migration Characterization
Rutherford Backscattering Spectroscopy (RBS)
RBS has emerged as a powerful, non-destructive technique for directly investigating elemental composition profiles and interface diffusion in perovskite devices [66]. The protocol involves:
Sample Preparation: Devices must be prepared and sealed in an inert environment (glove box under nitrogen) to prevent extrinsic degradation. Samples are typically stored for extended periods (up to one year) to study aged devices without external stress [66].
Measurement Conditions: RBS analysis is performed without applying potential or external stimulus. A beam of monoenergetic ions (typically alpha particles) is directed at the sample, and the energy distribution of backscattered ions is measured [66].
Data Analysis: The experimental spectra are fitted using specialized software (SIMNRA, XRUMP) to deconvolute contributions from different elements. The asymmetric shape of specific elemental spectra (I⁻, Br⁻, Cs⁺) indicates non-uniform concentration across film thickness, confirming migration [66].
Interpretation: Element-specific migration is identified through concentration depth profiles. For example, RBS has directly confirmed I⁻ as the most mobile species in CsPbI₂Br-based devices, followed by Cs⁺ ions, while Pb²⁺ shows negligible mobility [66].
4D Ultrafast Scanning Electron Microscopy (4D-USEM)
4D-USEM provides unprecedented spatiotemporal resolution for investigating surface defect dynamics and charge carrier behavior at the nanometer and femtosecond scales [69]. The methodology includes:
Experimental Setup: A photon-pump pulse (515 nm) excites the sample, synchronized with packets of pulsed electrons that probe the specimen's surface by collecting secondary electrons [69].
Surface Sensitivity: The technique selectively investigates the first few nanometers (≈5 nm) of the surface, where ionic migration and defect formation significantly influence device performance [69].
Dynamic Imaging: The photon-pump pulse induces contrast changes in illuminated areas, enabling real-space and real-time tracking of photoinduced charge carriers and their interaction with migrating ions [69].
Correlation with DFT: 4D-USEM results are complemented with density functional theory calculations to determine energy barriers for ion migration and validate experimental observations of surface vacancy passivation [69].
Drift-Diffusion-Poisson Modeling
Computational approaches provide valuable insights into ionic transport mechanisms and accumulation patterns under various operational conditions:
Model Framework: A semi-classical approach based on coupled drift-diffusion, Poisson, and continuity equations captures the dynamics of electrons, holes, mobile ions, and trap states [68].
Implementation: COMSOL Multiphysics simulations map ion migration and accumulation across the perovskite layer and interfaces under both transient and steady-state conditions [68].
Parameterization: Models incorporate temperature-dependent ion concentrations following Boltzmann statistics, with migration rates determined by activation energies specific to each ionic species [68].
Validation: Simulation results are compared with experimental measurements of current-voltage hysteresis, impedance spectroscopy, and direct elemental analysis to validate predictive accuracy [68].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Materials for Ionic Migration Studies
| Reagent/Material | Function in Research | Application Context |
|---|---|---|
| Trioctylphosphine oxide (TOPO) [60] | Surface passivation ligand | Coordinates with undercoordinated Pb²⁺ ions to suppress non-radiative recombination |
| Hexafluorophosphate (PF₆⁻) [56] | Multifunctional pseudo-halide anion | Passivates iodide vacancies, suppresses ion migration, provides hydrophobic barrier |
| l-Phenylalanine (L-PHE) [60] | Amino acid-based surface modifier | Enhances photostability through surface defect passivation |
| Reduced Graphene Oxide (RGO) [68] | Electrode material | Suppresses interfacial ion accumulation by order of magnitude vs. silver contacts |
| Formamidinium (FA) [69] | Large organic cation | Modulates iodide migration energy barriers for surface vacancy passivation |
Methodological Workflows and Causal Relationships
The following diagrams visualize key experimental workflows and the causal relationships between suppression strategies and device outcomes established in recent research.
Surface Modification and Characterization Workflow
Diagram 1: Comprehensive workflow for surface modification and characterization of perovskite quantum dots, integrating ligand engineering with advanced analytical techniques to establish correlations between surface chemistry and device performance.
Causal Relationships: Suppression Strategies to Device Outcomes
Diagram 2: Causal relationships between ionic migration suppression strategies and enhanced device outcomes, illustrating the multiple pathways through which surface and interface engineering improves both performance metrics and operational stability.
The comprehensive analysis presented in this whitepaper establishes an unequivocal correlation between suppressed ionic migration and enhanced device performance and lifetime in perovskite quantum dot systems. Through advanced characterization techniques and deliberate material design, researchers have demonstrated that targeted interventions at surfaces and interfaces can significantly mitigate the detrimental effects of ionic motion while simultaneously improving optoelectronic properties.
The most successful approaches share common characteristics: multi-functional design (simultaneously addressing multiple degradation pathways), energy barrier modulation (influencing migration kinetics), and interfacial optimization (preventing ion accumulation at critical junctions). The quantitative improvements summarized in this review—including PLQY enhancements of up to 18%, power conversion efficiencies exceeding 19% in PQD solar cells, and prolonged stability under continuous operation—provide compelling evidence for these strategies' efficacy.
Future research directions should prioritize the development of in operando characterization techniques capable of directly visualizing ionic migration dynamics under realistic operating conditions, the exploration of novel ligand chemistries specifically designed to create higher energy barriers for ionic motion, and the integration of machine learning approaches to identify optimal compositional combinations from the vast perovskite design space. As these efforts advance, the fundamental understanding of ionic migration dynamics in PQD surfaces will continue to enable increasingly sophisticated suppression strategies, ultimately unlocking the full commercial potential of perovskite-based optoelectronic devices.
Perovskite quantum dots (PQDs), particularly red-light-emitting variants, have garnered significant attention for their narrow-band emission and high color purity, which are ideal for advanced display technologies such as liquid crystal displays [34]. Despite their promising optical properties, the widespread practical application of red-light-emitting PQDs is primarily hindered by their inherent low photoluminescence quantum yield (PLQY) and susceptibility to degradation from environmental factors like moisture, heat, and light [34] [70]. Embedding PQDs within an inorganic glass matrix has emerged as an effective strategy to enhance their environmental durability. However, this encapsulation often results in reduced PLQY due to increased non-radiative recombination pathways [34].
Recent research has focused on elemental doping to simultaneously improve the stability and luminescent efficiency of PQDs. This case study examines the specific approach of doping CsPbBrI₂ PQD glass with silver iodide (AgI), a strategy that has demonstrated remarkable success in enhancing PLQY through interfacial mechanisms and the mitigation of ionic migration [34] [71]. The following analysis details the experimental protocols, characterizes the resulting enhancements, and elucidates the underlying mechanisms—particularly the role of localized surface plasmon resonance (LSPR) and bandgap modification—within the broader context of ionic migration dynamics in PQD surfaces.
Experimental Protocols
Synthesis of AgI-Doped CsPbBrI₂ PQD Glass
The synthesis of AgI-doped CsPbBrI₂ PQD glass involves a multi-stage process of melting, quenching, and heat treatment to precipitate quantum dots within an amorphous borosilicate glass matrix [34].
- Glass Composition: The base glass molar composition is 86 mol% (SiO₂-B₂O₃-ZnO-Na₂CO₃) and 14 mol% (Cs₂CO₃-PbBr₂-NaBr-PbI₂-NaI). Silver iodide (AgI) is added as a dopant at varying concentrations (X = 0, 0.1, 0.2, 0.4, and 0.6 mol%) [34].
- Melting and Quenching: A 10g batch of thoroughly ground raw materials is melted in a corundum crucible within a silicon molybdenum furnace at 1350°C for 30 minutes. The molten glass is rapidly quenched on a preheated copper mold to form a transparent, amorphous solid [34].
- Annealing and Heat Treatment: The obtained glass is immediately transferred to an annealing furnace held at 350°C for 3 hours to relieve internal stresses. Subsequently, the glass precursor is subjected to a controlled heat treatment at a temperature between 500°C and 540°C for 6 hours to precipitate CsPbBrI₂ PQDs and Ag nanoparticles (NPs) within the glass matrix [34] [72].
Characterization Techniques
A suite of characterization techniques is employed to analyze the structural, optical, and electronic properties of the synthesized composites.
- Structural Analysis: X-ray diffraction (XRD) identifies the crystal phase of the precipitated PQDs and confirms the presence of Ag nanoparticles [34].
- Optical Properties: UV-Vis spectroscopy determines the absorption characteristics and bandgap, while photoluminescence (PL) spectroscopy measures emission spectra, intensity, and full width at half maximum (FWHM). The photoluminescence quantum yield (PLQY) is quantitatively measured using an integrating sphere [34].
- Elemental and Morphological Analysis: Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) reveal the size, distribution, and lattice fringes of the PQDs and Ag NPs. Energy-dispersive X-ray spectroscopy (EDS) mapping confirms the elemental distribution within the glass composite [34].
- Theoretical Calculations: Density functional theory (DFT) calculations provide insights into electronic structure modifications, such as bandgap widening and changes in the density of states (DOS), induced by AgI doping [34].
Results and Discussion
Structural and Optical Properties
The incorporation of AgI dopants significantly influences the crystallization and optical behavior of CsPbBrI₂ PQDs.
- Enhanced Crystallization: XRD patterns demonstrate a gradual enhancement of the characteristic CsPbBrI₂ peaks with increasing AgI doping concentration, indicating that Ag⁺ ions promote the crystallization of perovskite quantum dots within the glass matrix [34].
- Optical Performance: The undoped CsPbBrI₂ PQD glass exhibits a PLQY of only 20%. With optimal AgI doping (0.4 mol%), the PLQY dramatically increases to 62.4% [34] [71]. The PL intensity initially increases with doping concentration up to 0.4 mol% but decreases at 0.6 mol%, indicating a concentration quenching effect.
Table 1: Summary of Optical Properties and Performance Metrics
| Property | Undoped PQD Glass | AgI-Doped (0.4 mol%) PQD Glass |
|---|---|---|
| PLQY | 20% | 62.4% |
| Bandgap | Baseline | Widened (DFT-calculated) |
| Stability (Intensity Retention) | Not Specified | >88% (under prolonged stress) |
| Emission Color | Red | Red (Narrow-band) |
Stability Enhancement
The AgI-doped PQD glass composite exhibits exceptional resilience under various stress conditions.
- Thermal, Water, and Photostability: The optimal sample (0.4 mol% AgI) maintains over 88% of its initial PL intensity under prolonged exposure to heat, moisture, and light, showcasing significantly improved durability compared to undoped counterparts [34].
- Synergistic Stabilization: The AgI doping contributes to a more stable crystal lattice, reducing halide vacancies and suppressing ionic migration, which is a primary cause of degradation in perovskite materials [34] [73].
Mechanisms of Enhancement
The dramatic improvement in PLQY and stability is attributed to two interconnected mechanisms.
- Localized Surface Plasmon Resonance (LSPR): The heat treatment process leads to the formation of Ag nanoparticles (NPs) within the glass matrix. Under light excitation, these Ag NPs exhibit LSPR, generating a strong localized electromagnetic field. This field interacts with the nearby PQDs, increasing their charge carrier density, radiative recombination rate, and overall emission efficiency—a phenomenon known as metal-enhanced fluorescence (MEF) [34].
- Bandgap Widening and Defect Passivation: DFT calculations confirm that AgI doping induces a widening of the PQD bandgap. This widening contributes to a reduction in non-radiative recombination pathways. Furthermore, the introduction of Ag⁺ ions triggers a charge compensation effect that facilitates ion migration (Cs⁺, Pb²⁺, Br⁻) during growth, promoting better crystallization and reducing the formation of non-radiative defect sites like halide vacancies [34].
The following diagram illustrates the experimental workflow and the key enhancement mechanisms involved in the creation of AgI-doped PQD glass.
The Scientist's Toolkit: Essential Research Reagents
The experimental synthesis relies on a specific set of high-purity chemical reagents, each serving a critical function in forming the glass matrix and the perovskite quantum dots.
Table 2: Key Research Reagent Solutions and Their Functions
| Reagent | Function in the Experiment |
|---|---|
| SiO₂, B₂O₃, ZnO, Na₂CO₃ | Forms the fundamental borosilicate glass network, providing mechanical strength and chemical inertness [34]. |
| Cs₂CO₃, PbBr₂, PbI₂ | Principal precursors for the CsPbBrI₂ perovskite quantum dots. Provides Cs⁺, Pb²⁺, Br⁻, and I⁻ ions [34]. |
| NaBr, NaI | Additional halide sources to adjust and balance the composition of the perovskite [34]. |
| AgI | Dopant precursor. Source of Ag⁺ ions, which facilitate the formation of Ag NPs and modify the electronic structure of the PQDs [34]. |
| Al₂O₃ Crucible | High-temperature container for melting the glass batch, chosen for its thermal stability and resistance to chemical reaction [34]. |
This case study demonstrates that doping with AgI is a highly effective strategy for enhancing the performance of red-emitting CsPbBrI₂ PQDs in glass. The key achievement is a more than three-fold increase in PLQY, reaching 62.4%, coupled with exceptional operational stability. The underlying mechanisms—LSPR from in-situ formed Ag nanoparticles and AgI-induced bandgap widening and defect reduction—work synergistically to enhance radiative recombination and suppress non-radiative pathways. These findings provide profound insights into managing ionic migration and surface dynamics in PQDs, paving the way for their application in high-performance, long-lasting optoelectronic devices such as wide-color-gamut displays.
Biocompatibility and Toxicity Assessments for Biomedical Readiness
The integration of advanced materials, such as perovskite quantum dots (PQDs), into biomedical devices represents a frontier in diagnostic and therapeutic technology. The core thesis of this whitepaper is that the ionic migration dynamics inherent to PQD surfaces are not merely a materials science phenomenon but a central determinant of their biological safety and performance. Biocompatibility—the ability of a material to perform with an appropriate host response in a specific application—is a fundamental requirement for regulatory approval and clinical success. For PQDs, whose ionic lattice structure facilitates the migration of ions like lead (Pb²⁺), iodide (I⁻), and bromide (Br⁻) under physiological conditions, traditional biocompatibility assessment frameworks require augmentation. This document provides an in-depth technical guide for researchers and drug development professionals, framing classical toxicity endpoints within the critical context of PQD surface ion dynamics. It outlines how these dynamics influence degradation profiles, ion release kinetics, and ultimately, the biological response, thereby establishing a roadmap for achieving biomedical readiness.
Regulatory Framework and Essential Biocompatibility Endpoints
Biocompatibility assessment is a risk-based process governed by internationally recognized standards, primarily the ISO 10993 series, and mandated by regulatory bodies like the U.S. Food and Drug Administration (FDA) and the European Union's Medical Device Regulation (MDR) [74]. The evaluation required for a device is determined by its nature of body contact and the contact duration [75].
- Nature of Body Contact: Devices are categorized as surface devices (e.g., contacting skin or mucosal membranes), externally communicating devices (e.g., contacting blood path indirectly or tissue/bone), or implant devices. Each category necessitates a different set of biological evaluations.
- Contact Duration: This is classified as Limited (≤24 hours), Prolonged (24 hours to 30 days), or Long-term/Permanent (>30 days). Longer and more invasive contact typically requires more extensive testing [75].
The "Big Three" biocompatibility tests—cytotoxicity, sensitization, and irritation—are required for almost all medical devices, regardless of category or contact duration [74]. However, as detailed in Table 1, devices with more invasive and prolonged contact require assessment of additional endpoints like systemic toxicity, genotoxicity, hemocompatibility, and implantation effects [75].
Table 1: Biocompatibility Evaluation Endpoints by Device Category and Contact Duration
| Biological Effect | Device Category | Limited | Prolonged | Long-term |
|---|---|---|---|---|
| Cytotoxicity | All Categories | |||
| Sensitization | All Categories | |||
| Irritation | All Categories | |||
| Acute Systemic Toxicity | Mucosal Membrane, Breached Surface, Blood Path, Tissue/Bone, Implants | |||
| Genotoxicity | Breached Surface, Blood Path, Tissue/Bone, Implants | |||
| Hemocompatibility | Blood Path, Circulating Blood, Implant Device: Blood | |||
| Implantation | Tissue/Bone, Blood | |||
| Carcinogenicity | Breached Surface, Blood Path, Tissue/Bone, Implants |
For PQD-based devices, a pivotal additional consideration is degradation information, which must be provided for any device or material intended to degrade or from which ions may be released [75] [76]. The by-products of degradation, including migrated ions, must be assessed for their toxicological profile.
The "Big Three" Tests: Protocols and PQD-Specific Considerations
Cytotoxicity Testing
Purpose: To determine if a device or its extracts cause cell death or damage.
- Protocol (ISO 10993-5): Extracts of the PQD device are prepared using appropriate solvents like physiological saline or cell culture medium, often incubated at 37°C for 24 hours [74] [75]. This extract is then applied to mammalian cell lines, such as L929 fibroblasts or Vero cells, for approximately 24 hours [74].
- Assessment Endpoints: Cell viability is quantitatively measured using assays like MTT, XTT, or Neutral Red Uptake, which measure metabolic activity [74]. Morphological changes, cell detachment, and lysis are also evaluated microscopically.
- PQD-Specific Considerations: The extract preparation is a critical step for PQDs, as it simulates the release of ions. The high ionic mobility of lead-based PQDs means that Pb²⁺ release can be a significant cytotoxic driver [13]. Testing should use extract conditions that reflect the ionic strength and pH of the physiological environment to accurately model ion migration and release. Cell viability above 70% is generally considered a positive sign for neat extract [74].
Sensitization Testing
Purpose: To evaluate the potential for a device to cause an allergic reaction (delayed-type hypersensitivity).
- Protocol (ISO 10993-10): The Guinea Pig Maximization Test (GPMT) or Local Lymph Node Assay (LLNA) are traditional animal models. Increasingly, in vitro methods based on the activation of dendritic cell-like lines or peptide reactivity assays are being developed [74].
- PQD-Specific Considerations: The released ions or degraded by-products from PQDs can act as haptens, binding to skin proteins and potentially triggering an immune response. The assessment must evaluate not the PQD material itself, but the potential for its released components to cause sensitization.
Irritation or Intracutaneous Reactivity Testing
Purpose: To determine if a device or its extracts cause localized, reversible inflammatory responses.
- Protocol (ISO 10993-23): Extracts are injected intradermally into rabbits (intracutaneous reactivity) or applied to skin models (e.g., reconstructed human epidermis, EpiDerm) for irritation. The test and control sites are scored for erythema and edema.
- PQD-Specific Considerations: The ionic release from PQDs can alter the pH and osmolarity of the extract, which are key factors in irritation potential. These parameters must be measured and controlled for in the test system.
The following workflow visualizes the standard process for initiating these core biocompatibility tests for a PQD-based device.
Material Degradation and Ionic Release Assessment
For PQDs, the assessment of material degradation is inseparable from the evaluation of ionic migration. The ionic crystal lattice of perovskites is dynamic, and ions can migrate under the influence of internal or external electric fields, concentration gradients, and moisture [77] [78]. This migration directly influences what ions are released into the biological environment and at what rate.
- Degradation Mechanisms: PQD degradation in aqueous physiological environments can occur through hydrolysis, where water molecules attack the ionic bonds in the crystal lattice [76]. This process is accelerated by the migration of halide ions (I⁻, Br⁻) to the surface, creating defects and initiating the dissolution of the structure and release of Pb²⁺.
- Ionic Migration Dynamics: Studies on perovskite solar cells and detectors have shown that under an electric field, iodine ions (I⁻) are highly mobile and can accumulate at interfaces, forming carrier transport barriers and modifying band-bending [77] [78]. In a biological context, this translates to a mechanism for rapid ion release and localized accumulation. The activation energy for ion migration ((E_{a}^{ion})) is a key parameter that can be fitted from electrical measurements to understand and predict migration rates [78].
Table 2: Key Analytical Techniques for Assessing PQD Degradation and Ion Release
| Assessment Approach | Specific Techniques | Measured Parameters | Relevance to PQDs |
|---|---|---|---|
| Physical | Gravimetric Analysis (Mass loss) | Weight loss over time in simulated body fluid. | Infers dissolution but cannot distinguish ion release from particle detachment [76]. |
| Scanning Electron Microscopy (SEM) | Surface morphology, cracking, erosion. | Visualizes physical degradation onset and defect formation from ion migration [76]. | |
| Chemical | Inductively Coupled Plasma Mass Spectrometry (ICP-MS) | Quantifies released metal ions (e.g., Pb²⁺, Cs⁺) in solution. | Gold standard for directly measuring toxic ion release kinetics [13]. |
| Nuclear Magnetic Resonance (NMR) / High-Performance Liquid Chromatography (HPLC) | Identifies and quantifies organic by-products (e.g., ligands). | Tracks stability of surface capping ligands that suppress ion release [76]. | |
| Fourier-Transform Infrared Spectroscopy (FTIR) | Tracks chemical bond changes (e.g., hydrolysis of esters). | Monitors degradation of surface organic ligands [76]. | |
| Indirect (Functional) | Surface Zeta Potential (SZP) Measurement | Electrostatic charge at material-solution interface. | Highly negative SZP (e.g., -90 mV) indicates strong ion depletion zone formation, repelling co-ions [49]. Useful for filtration applications. |
The diagram below illustrates the interconnected cycle of ionic migration, material degradation, and biological response that forms the core challenge in assessing PQD biocompatibility.
Mitigation Strategies and Lead-Free Compositions
The primary toxicity concern for lead-halide PQDs is the release of Pb²⁺ ions, which are known systemic toxicants. Therefore, mitigation strategies are a critical component of biomedical readiness.
- Surface Passivation and Ligand Engineering: A primary strategy involves engineering a robust, conductive capping layer on the PQD surface to "lock" ions in the lattice. Recent advances include an Alkali-Augmented Antisolvent Hydrolysis (AAAH) strategy, which uses alkaline environments to facilitate rapid substitution of pristine insulating ligands with conductive counterparts, creating a denser and more stable capping layer [14]. This has been shown to reduce trap-states and improve stability, thereby reducing the pathways for ion escape.
- Lead-Free Perovskite Alternatives: The most direct mitigation strategy is to eliminate lead entirely. Bismuth-based PQDs (e.g., Cs₃Bi₂Br₉) are emerging as promising candidates. Research shows they offer extended serum stability and, critically, their composition means they already meet current safety standards without requiring additional coating for lead containment [13]. While their optoelectronic properties may differ, they present a significantly de-risked pathway for biomedical applications.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for PQD Biocompatibility and Ionic Migration Research
| Reagent/Material | Function in Research | Technical Notes |
|---|---|---|
| L929 Fibroblast Cell Line | In vitro model for cytotoxicity testing (ISO 10993-5). | Sensitive indicator of leachate toxicity; used with MTT/XTT assays [74]. |
| Reconstituted Human Epidermis (EpiDerm) | In vitro model for skin irritation testing. | Replaces rabbit testing for irritation; assesses response to PQD extracts [74]. |
| Methyl Benzoate (MeBz) Antisolvent | Used in ligand exchange for surface passivation. | In AAAH strategy, its hydrolysis products form stable, conductive capping on PQDs [14]. |
| Potassium Hydroxide (KOH) | Creates alkaline environment for antisolvent hydrolysis. | Facilitates spontaneous ester hydrolysis, enabling dense conductive ligand capping on PQDs [14]. |
| Phosphate Buffered Saline (PBS) | Standard medium for device extraction and degradation studies. | Simulates ionic strength of physiological fluids; medium for studying ion release kinetics [76]. |
| Poly(allylamine hydrochloride) (PAH) / Polystyrene sulfonate (PSS) | Polyelectrolytes for layer-by-layer surface charge control. | Used to create membranes with controlled Surface Zeta Potential (SZP) to model ion-depletion zones [49]. |
| Cs₃Bi₂Br₉ PQD Precursors | Synthesis of lead-free, bismuth-based PQDs. | Key for developing inherently safer PQD formulations with reduced toxicity concerns [13]. |
Achieving biomedical readiness for technologies based on perovskite quantum dots demands a sophisticated, two-pronged approach. First, it requires strict adherence to the established regulatory framework of biocompatibility assessment, as defined by ISO 10993 and relevant regulatory bodies. The "Big Three" tests—cytotoxicity, sensitization, and irritation—form the non-negotiable foundation of this assessment. Second, and more critically, it necessitates a deep investigation into the ionic migration dynamics unique to the PQD material class. The release of ions, particularly lead, driven by migration and subsequent material degradation, is the primary safety hurdle. Future progress hinges on the development and standardization of specific degradation and ion release assays, the adoption of robust surface passivation strategies like the AAAH approach, and a concerted shift towards the exploration and optimization of lead-free alternatives such as bismuth-based PQDs. By integrating classical biocompatibility protocols with a fundamental understanding of material-specific ion dynamics, researchers can successfully navigate the path from laboratory innovation to safe clinical application.
Conclusion
The precise understanding and control of ionic migration dynamics on PQD surfaces are paramount for transitioning from laboratory curiosities to reliable biomedical tools. This synthesis of knowledge confirms that strategic surface engineering, through advanced ligand chemistry and targeted doping, can effectively suppress detrimental ion movement, leading to dramatic improvements in PQD stability and optoelectronic performance. The convergence of high-resolution characterization techniques and theoretical modeling is paving the way for predictive design. Future directions must focus on establishing standardized protocols for quantifying ionic migration under biologically relevant conditions, developing novel biocompatible ionic liquid formulations for enhanced drug permeability, and conducting rigorous in vivo validation studies. Mastering these surface ion dynamics will ultimately unlock the full potential of PQDs in creating innovative, non-invasive therapeutic and diagnostic platforms.
References
- 1. Toolsets for assessing ionic migration in halide perovskites [https://www.sciencedirect.com/science/article/pii/S2542435124001065]
- 2. Non-contact measurement of ion mobility in single halide ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04296h?page=search]
- 3. Quantification of light-enhanced ionic transport in lead ... [https://www.nature.com/articles/lsa2016243]
- 4. Ion Migration and Interface Engineering in Emerging ... [https://www.sciencedirect.com/science/article/pii/S2468217925001674]
- 5. Benchmarking machine learning models for predicting ... [https://www.nature.com/articles/s41524-025-01571-z]
- 6. Modeling phase transformations in Mn-rich disordered ... [https://arxiv.org/html/2506.20605v2]
- 7. Cation Exchange in Lead Halide Perovskite Quantum Dots ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11935038/]
- 8. How to improve the structural stabilities of halide perovskite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10821855/]
- 9. Thermal tolerance of perovskite quantum dots dependent ... [https://www.nature.com/articles/s41467-023-37943-6]
- 10. Enhancing the stability of red-emitting CsPbI x Br 3−x QDs for ... [https://pubs.rsc.org/ko/content/articlehtml/2025/nr/d5nr03199k]
- 11. Ligand Engineering of Inorganic Lead Halide Perovskite ... [https://www.mdpi.com/2079-4991/14/14/1201]
- 12. Pioneering perovskite quantum dot nanosensors for heavy ... [https://www.sciencedirect.com/org/science/article/pii/S2046206925030244]
- 13. Next-Generation Biosensing with Perovskite Quantum Dots [https://www.sciencedirect.com/science/article/pii/S1872204025001860]
- 14. Enriching conductive capping by alkaline treatment of ... [https://www.nature.com/articles/s41467-025-63618-5]
- 15. Advancement of technology towards developing perovskite ... [https://www.nature.com/articles/s44296-025-00073-9]
- 16. Advances in Perovskite Quantum Dot Optoelectronics [https://insights.pluto.im/advances-in-perovskite-quantum-dot-optoelectronics-towards-high-performance-leds-and-solar-cells/]
- 17. Halide Perovskite Nanostructures: Processing Methods ... [https://www.nature.com/articles/s44310-025-00090-5]
- 18. Surface Traps in Colloidal Quantum Dots: A Combined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5651579/]
- 19. Leveraging transfer learning for accurate estimation of ... [https://arxiv.org/html/2508.06436v1]
- 20. Reconfiguring surface functions using visible-light ... [https://www.nature.com/articles/s41467-018-06180-7]
- 21. Electrochemical modulation enhances the selectivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9191649/]
- 22. Visualization of lithium-ion transport and phase evolution ... [https://www.nature.com/articles/ncomms15400]
- 23. How to improve the structural stabilities of halide perovskite ... [https://nanoconvergencejournal.springeropen.com/articles/10.1186/s40580-024-00412-x]
- 24. Ion Mobility Spectrometry: Fundamental Concepts ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6832852/]
- 25. Guidelines to correctly measure the lithium ion conductivity ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775322003366]
- 26. Precision Spectroscopy and Quantum Control of Trapped ... [https://www.nist.gov/programs-projects/precision-spectroscopy-and-quantum-control-trapped-molecular-ions]
- 27. Toolsets for assessing ionic migration in halide perovskites - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S2542435124001065]
- 28. Extension of microscale surface ion conduction (μSIC) to the ... [https://pubs.rsc.org/en/content/articlehtml/2025/an/d5an00584a]
- 29. Robust skin-integrated conductive biogel for high-fidelity ... [https://www.nature.com/articles/s41467-024-55417-1]
- 30. Synergetic effect of ligand removal, reparation, and surface ... [https://www.sciencedirect.com/science/article/abs/pii/S246802302200863X]
- 31. Room Temperature Ionic Liquid Capping Layer for High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11109663/]
- 32. Molecular dynamics simulations of ion adsorption on ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433224029829]
- 33. Overcoming the luminescence instability of colloidal mixed-halide perovskite quantum dots through ion motion confinement - Journal of Materials Chemistry C (RSC Publishing) [https://pubs.rsc.org/en/content/articlelanding/2024/tc/d4tc01279h]
- 34. Interfacial mechanisms of enhanced photoluminescence in ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979725000761]
- 35. Effect of Surface Ligand Modification on the Optical ... [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5480005]
- 36. Photophysics and photovoltaic properties of Zn-alloyed Ag- ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838822026871]
- 37. Exploration of dopant and surface passivation on optical ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838819306474]
- 38. “Popping the Ion‐Basket”: Enhancing Thermoelectric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11924034/]
- 39. Quantifying and Reducing Ion Migration in Metal Halide ... [https://www.mdpi.com/1420-3049/28/13/5026]
- 40. Ionic liquid-based transdermal drug delivery systems for ... [https://pubs.rsc.org/en/content/articlelanding/2025/cc/d5cc03935e]
- 41. Transdermal drug delivery system for biopharmaceuticals ... [https://kyushu-u.elsevierpure.com/en/publications/transdermal-drug-delivery-system-for-biopharmaceuticals-using-ion/]
- 42. Ionic Liquid-Based Immunization Patch for the Transdermal ... [https://www.mdpi.com/1420-3049/29/13/2995]
- 43. Ionic liquids self-assembled micelles for transdermal delivery [https://pubmed.ncbi.nlm.nih.gov/40688029/]
- 44. An ionic liquid nanoemulsion transdermal delivery system ... [https://pubmed.ncbi.nlm.nih.gov/40975175/]
- 45. Electrically induced directional ion migration in two- ... [https://www.sciencedirect.com/science/article/pii/S2590238524001127]
- 46. Suppressing non-radiative recombination for efficient and ... [https://pubs.rsc.org/en/content/articlehtml/2025/ee/d4ee02917h]
- 47. Understanding ion migration suppression in all-inorganic ... [https://www.sciopen.com/article/10.26599/NRE.2025.9120166]
- 48. Rational regulation strategies toward perovskite precursor ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta01221j]
- 49. Migration of ions near charged surface - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8081195/]
- 50. Deterministic resonance fluorescence improvement of ... [https://www.nature.com/articles/s41377-025-01838-6]
- 51. Top-Down Dual-Interface Carrier Management for Highly ... [https://link.springer.com/article/10.1007/s40820-024-01631-x]
- 52. Balancing Molecular Sensitization and Surface Passivation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616780/]
- 53. Events for November 2025 - Penn Engineering Events | [https://events.seas.upenn.edu/event/mse-david-p-pope-distinguished-lecture-ion-migration-and-its-impact-on-the-stability-of-halide-perovskite-solar-cells-prashant-kamat-university-of-notre-dame/?utm_source=rss&utm_medium=rss&utm_campaign=mse-david-p-pope-distinguished-lecture-ion-migration-and-its-impact-on-the-stability-of-halide-perovskite-solar-cells-prashant-kamat-university-of-notre-dame]
- 54. Ligand engineering of perovskite quantum dots for efficient ... [https://www.sciencedirect.com/science/article/abs/pii/S2095495622000730]
- 55. Thermal tolerance of perovskite quantum dots dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10113222/]
- 56. Fluorinated Pseudo-Halide Anion Enables >19% Efficiency ... [https://pubmed.ncbi.nlm.nih.gov/40817595/]
- 57. Consecutive surface matrix engineering of FAPbI3 ... [https://pubs.rsc.org/en/content/articlelanding/2025/ee/d5ee02127h]
- 58. Dual-Phase Ligand Engineering Enables 18.21% FAPbI3 ... [https://ui.adsabs.harvard.edu/abs/2025AdM....3717346L/abstract]
- 59. Enriching conductive capping by alkaline treatment of ... [https://pubmed.ncbi.nlm.nih.gov/41022725/]
- 60. Effect of Surface Ligand Modification on the Optical ... [https://www.sciencedirect.com/science/article/abs/pii/S2468023025023211]
- 61. Strategies for performance and stability advancement in ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta04375a]
- 62. Optimum temperature of atomic layer deposition of alumina ... [https://www.sciencedirect.com/science/article/abs/pii/S0022231323002387]
- 63. Studying ion transport dynamics in electrochemical ... [https://www.sciencedirect.com/science/article/pii/S1572665724003771]
- 64. Ultrasensitive dopamine detection using CsPbBr3-PQD-COF ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12136283/]
- 65. Increase in Interfacial Adhesion and Electrochemical ... [https://www.nature.com/articles/s41598-019-38615-6]
- 66. Experimental evidence of ion migration in aged inorganic ... [https://pubs.rsc.org/en/content/articlehtml/2022/ma/d2ma00199c]
- 67. Ion Migration in Metal Halide Perovskites: Characterization ... [https://pubmed.ncbi.nlm.nih.gov/38941623/]
- 68. Simulation of ion migration across the structure ... [https://www.sciencedirect.com/science/article/abs/pii/S0921510725008104]
- 69. Mapping Surface‐Defect and Ions Migration in Mixed‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11516060/]
- 70. Effects of Tellurium Doping on Environmental Stability and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9245131/]
- 71. Interfacial mechanisms of enhanced photoluminescence in ... [https://pubmed.ncbi.nlm.nih.gov/39809024/]
- 72. Synergistic enhancement of the stability of CsPbBrI 2 ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825039957]
- 73. Understanding the influence of cation and anion migration ... [https://www.nature.com/articles/s41598-023-42933-1]
- 74. The “Big Three” in biocompatibility testing of medical devices [https://pmc.ncbi.nlm.nih.gov/articles/PMC10800850/]
- 75. Biocompatibility Evaluation Endpoints by Device Category [https://www.fda.gov/medical-devices/biocompatibility-assessment-resource-center/biocompatibility-evaluation-endpoints-device-category]
- 76. A review of biomaterial degradation assessment ... [https://www.nature.com/articles/s41529-024-00487-1]
- 77. Ionic migration induced loss analysis of perovskite solar cells [https://pubs.rsc.org/en/content/articlelanding/2022/cp/d1cp05450c]
- 78. Study of electric-field induced ionic migration on all ... [https://ui.adsabs.harvard.edu/abs/2024JAP...135c5304Z/abstract]