Beyond the Spacer: How Surface Ligand Engineering Dictates Charge Transport in Quantum Dot Solids
This article comprehensively explores the critical role of surface ligand engineering in controlling charge transport within quantum dot (QD) solids, a key parameter for developing high-performance optoelectronic and biomedical devices.
Beyond the Spacer: How Surface Ligand Engineering Dictates Charge Transport in Quantum Dot Solids
Abstract
This article comprehensively explores the critical role of surface ligand engineering in controlling charge transport within quantum dot (QD) solids, a key parameter for developing high-performance optoelectronic and biomedical devices. We first establish the foundational principles, explaining how ligand length influences quantum confinement, inter-dot coupling, and the transition from insulating to conductive behavior. The discussion then progresses to methodological strategies, including ligand exchange processes and the innovative use of redox-active and hybrid ligands to actively participate in charge transport. Furthermore, we address central challenges such as managing energetic disorder, mitigating surface defects, and optimizing solid-state packing. Finally, the article covers advanced validation techniques, from ultrafast spectroscopy to electrochemical and device-level characterization, that correlate ligand structure with device performance. This review serves as a strategic guide for researchers aiming to rationally design QD solids with tailored charge transport properties for advanced applications.
The Fundamental Principles: How Ligand Length Governs Inter-Dot Coupling and Confinement
Quantum dot (QD) solids are materials formed through the assembly of individual nanocrystals into dense, solid films. These materials exhibit unique electronic properties that arise not only from the quantum confinement effect within each dot but also from the interparticle coupling between them [1]. When QD size is reduced to or below the Bohr exciton radius, charge carriers become confined within the structural boundaries, leading to size-tunable optical and electronic properties [2]. This tunability makes QD solids promising building blocks for next-generation technologies including photovoltaics, photodetectors, light-emitting diodes (LEDs), and thermoelectrics [2] [1]. The surface chemistry of these quantum dots, governed by their ligand shells, plays a critical role in determining the charge transport properties of the resulting solids. Through careful engineering of surface ligands, researchers can modulate everything from interparticle spacing to electronic structure, enabling precise control over material performance in energy applications [3].
Fundamental Mechanisms of Charge Transport in QD Solids
Electronic Transport Mechanisms
Charge transport in QD solids occurs through several distinct mechanisms, with the dominant pathway heavily influenced by surface ligand chemistry:
- Band-like conduction occurs when strong electronic coupling between neighboring QDs allows carriers to delocalize across multiple dots, resembling transport in bulk semiconductors. This requires very short interparticle distances typically achieved with minimal ligand barriers [1].
- Hopping transport represents the more common mechanism where carriers tunnel between localized states on individual QDs. This process is highly sensitive to interdot distance, following an exponential relationship: shorter ligands reduce tunneling barriers and significantly increase hopping rates [4] [1].
- Tunneling through ligand barriers represents another pathway where carriers penetrate through the energy barrier presented by the organic ligand shell [1].
A groundbreaking shift from traditional thinking has emerged with the concept of "active" versus "passive" ligand roles. Most conventional ligands act as passive spacers whose primary function is to modulate interparticle distance. In contrast, redox-active ligands introduce electronic states that provide an additional pathway for charge transport via self-exchange chain reactions [4]. This represents a paradigm shift in ligand design, where ligands no longer merely get "out of the way" of charge transport but actively participate in the process.
Table 1: Charge Transport Mechanisms in Quantum Dot Solids
| Mechanism | Description | Key Influencing Factors | Role of Ligands |
|---|---|---|---|
| Band-like Conduction | Delocalized carriers moving through strongly coupled QDs | Interdot electronic coupling, energetic disorder | Minimize barrier thickness to enable wavefunction overlap |
| Hopping Transport | Thermal-assisted tunneling between localized states | Interparticle distance, temperature, energy level alignment | Determine tunneling distance and barrier height |
| Tunneling | Quantum mechanical penetration through barriers | Barrier height and width | Act as the potential barrier carriers must tunnel through |
| Redox-Mediated Transport | Charge transfer via active ligand states | Ligand redox potential, coverage density, ion transport | Provide active electronic states for charge hopping |
The Critical Role of Surface Ligands
Surface ligands fundamentally determine charge transport efficiency in QD solids through multiple interconnected mechanisms:
- Interparticle Spacing: Ligands form physical barriers between QDs. Longer aliphatic chains (e.g., oleic acid) maintain large separations (>2 nm) that severely limit electronic coupling, while shorter ligands (e.g., halides, pyridine) reduce this distance to <1 nm, dramatically increasing carrier mobility [5] [1].
- Trap State Passivation: Undercoordinated atoms on QD surfaces create electronic trap states that non-radiatively recombine charge carriers. Properly matched ligands (e.g., halides for Pb atoms) coordinate with these sites, reducing trap density and increasing photoluminescence quantum yield [5] [3].
- Electronic Coupling: Certain ligands, particularly conjugated molecules and redox-active species, actively mediate electronic interactions between QDs by providing orbital pathways for charge delocalization [4] [1].
- Doping and Band Alignment: Ligands can introduce doping effects by altering surface stoichiometry or introducing charge impurities. For instance, specific ligands can shift the Fermi level position, enabling n-type or p-type behavior [3] [6].
Diagram 1: Ligand Engineering Workflow for Quantum Dot Solids. This flowchart illustrates the decision process for selecting ligand types based on desired material properties.
Experimental Methods for Probing Charge Transport
Advanced Spectroscopic Techniques
Understanding charge dynamics in QD solids requires specialized characterization methods capable of probing ultrafast processes:
Resonant Auger Spectroscopy (RAS) and Core-Hole Clock Spectroscopy (CHCS) employ the core-hole lifetime as an internal clock to measure attosecond-scale electron transfer dynamics [2]. When X-ray excitation creates a transient core-hole state, the resonantly excited electron can either remain localized (Raman decay) or delocalize (Auger decay) within the core-hole lifetime (0.26 fs for Pb 3d). The intensity ratio between these decay channels provides quantitative charge transfer times, revealing that larger PbS QDs and bulk samples exhibit faster charge transfer compared to smaller dots [2].
Fluorescence Lifetime Imaging Microscopy (FLIM) spatially maps charge transfer rates at QD-microbe interfaces by tracking photoluminescence decay [7]. This technique revealed two distinct electron transfer mechanisms in CdSe QD-microbe systems: a faster pathway (1.5×10⁹ s⁻¹) with fewer acceptors and a slower pathway (4.1×10⁸ s⁻¹) with more acceptors, assigned to indirect and direct transfer mechanisms respectively [7].
Electrochemical Spectroscopy using cyclic voltammetry characterizes charge transfer processes in QD/redox ligand assemblies by scanning the Fermi level while monitoring current [4]. This approach identifies distinct signals corresponding to charge injection into the QD conduction band and electron transfer through redox ligand states.
Device-Level Characterization
Field-effect transistors (FETs) provide a platform for evaluating charge transport properties under controlled conditions. Studies on InP QDs with hybrid S²⁻/N³⁻ ligand exchange demonstrated electron mobilities of 0.45 cm² V⁻¹ s⁻¹, approximately 10 times higher than previous reports [6]. This improvement resulted from enhanced interdot coupling and reduced trap-state densities.
Photovoltaic device characterization reveals how ligand engineering impacts solar cell performance. PbS QD solar cells employing hybrid halide/pyridine passivation achieved power conversion efficiencies of 6.8% compared to 5.3% for halide-only treated cells, with improvements in all device parameters (Jsc, Voc, FF) due to better surface passivation and charge extraction [5].
Table 2: Experimental Techniques for Studying Charge Transport in QD Solids
| Technique | Physical Principle | Timescale Resolution | Key Measurable Parameters |
|---|---|---|---|
| Core-Hole Clock Spectroscopy | Core-hole lifetime as internal clock | Attoseconds (10⁻¹⁸ s) | Charge transfer times, electron delocalization rates |
| Fluorescence Lifetime Imaging | Photoluminescence decay dynamics | Picoseconds to nanoseconds (10⁻¹²-10⁻⁹ s) | Charge transfer rates, acceptor densities, spatial heterogeneity |
| Cyclic Voltammetry | Current response to potential sweep | Milliseconds to seconds (10⁻³-10⁰ s) | Redox potentials, charge injection rates, transport mechanisms |
| Field-Effect Transistor | Gate-modulated conductivity | Steady-state to microseconds | Carrier mobility, carrier density, trap states |
| Transient Absorption | Pump-probe carrier dynamics | Femtoseconds to nanoseconds (10⁻¹⁵-10⁻⁹ s) | Carrier lifetimes, recombination rates, trap states |
Ligand Engineering Strategies and Protocols
Ligand Exchange Methodologies
Successful ligand engineering requires precise control over the replacement of native synthetic ligands with functional counterparts:
Solution-Phase Exchange involves exposing QDs to excess new ligands in solution before film formation. This approach typically achieves more complete exchange but risks colloidal instability. For InP QDs, solution-based exchange with S²⁻ enabled effective removal of native ligands while controlling n-doping [6].
Solid-State Exchange employs ligand solutions to treat pre-formed QD films, preserving film integrity while modifying surface chemistry. Sequential treatment with tetrabutylammonium iodide (TBAI) and pyridine on PbS QD films created hybrid-passivated systems with improved morphology and reduced cracking [5].
Hybrid Multistep Exchanges combine both approaches for optimal results. For InP QDs, researchers developed a strategy with solution-based S²⁻ exchange followed by solid-state N³⁻ treatment, further enhanced by selenium capping to achieve record mobilities [6].
Ligand Classification and Selection
- Short Organic Ligands: Molecules like pyridine and ethanedithiol reduce interparticle distance while providing moderate passivation. Their small size enables tight QD packing but may offer incomplete surface coverage [5].
- Inorganic Ligands: Halide ions (I⁻, Br⁻, Cl⁻) and chalcogenides (S²⁻, Se²⁻) provide strong metal coordination and excellent electronic passivation. In PbS QDs, iodine ions effectively coordinate with Pb atoms, reducing trap states and enhancing conductivity [5] [6].
- Redox-Active Ligands: Molecules like ferrocene carboxylate introduce accessible electronic states that actively mediate charge transport through self-exchange reactions [4]. In ZnO-FcCOO⁻ assemblies, this created two complementary transport pathways: QD band conduction and ligand-mediated hopping.
- Hybrid Ligand Systems: Combinations of different ligand types address multiple requirements simultaneously. The TBAI-pyridine hybrid on PbS QDs enabled both excellent passivation (from pyridine) and high charge mobility (from iodide) [5].
Diagram 2: Charge Transport Pathways Mediated by Different Ligand Types. This diagram illustrates how various ligand classes facilitate or hinder electron transfer between quantum dots.
Research Reagent Solutions: Essential Materials for Ligand Engineering
Table 3: Key Reagents for Quantum Dot Surface Engineering
| Reagent/Chemical | Function | Application Example | Effect on Charge Transport |
|---|---|---|---|
| Tetrabutylammonium Iodide (TBAI) | Halide source for inorganic passivation | PbS QD solar cells [5] | Enhances carrier mobility, reduces trap states |
| Pyridine | Short organic ligand for distance reduction | Hybrid passivation of PbS QDs [5] | Decreases interdot distance, improves packing |
| Ferrocene Carboxylic Acid | Redox-active ligand for active transport | ZnO QD assemblies [4] | Enables self-exchange chain reaction transport |
| Ammonium Sulfide | Inorganic sulfide source for passivation | InP QD field-effect transistors [6] | Enhances interdot coupling, controls doping |
| Sodium Azide | Nitrogen source for surface modification | InP QD thin films [6] | Further reduces trap states, improves mobility |
| Selenium Powder | Chalcogenide source for surface capping | InP QD device optimization [6] | Increases carrier diffusion length, reduces traps |
Impact on Device Performance and Applications
Photovoltaic Devices
In quantum dot solar cells, ligand engineering directly impacts all key performance parameters. PbS QD solar cells employing hybrid TBAI-pyridine passivation demonstrated remarkable improvements: power conversion efficiency increased from 5.3% to 6.8%, attributed to better surface passivation, reduced trap-assisted recombination, and enhanced charge extraction [5]. The ligand exchange process critically influences the open-circuit voltage (Voc) deficit relative to the bandgap, with insufficient passivation leading to sub-bandgap states that limit Voc [5].
For CsPbI3 perovskite QD solar cells, a innovative "solvent-mediated ligand exchange" approach enabled exceptional 16.53% efficiency - a record for inorganic QD photovoltaics at the time [8]. By optimizing the solvent environment for ligand substitution, researchers achieved more complete replacement of insulating native ligands while maintaining QD structural integrity, significantly reducing defect density and improving carrier mobility.
Electronic and Optoelectronic Devices
Field-effect transistors serve as sensitive probes of charge transport in QD solids. InP QD FETs with hybrid S²⁻/N³⁻ ligand exchange and Se capping demonstrated electron mobilities of 0.45 cm² V⁻¹ s⁻¹, approximately 10× higher than previous reports, with on-off current ratios of 10³-10⁴ [6]. These improvements stemmed from lower trap-state densities and longer carrier lifetimes, yielding a four-fold increase in carrier diffusion length.
In nano-bio hybrid systems, ligand engineering enables charge transfer between QDs and biological entities. CdSe QDs with BF4⁻ surface moieties transferred electrons to Shewanella oneidensis microbes at rates of 10⁸ to 10¹⁰ s⁻¹, with the ligand shell determining the proximity requirements for direct versus indirect electron transfer mechanisms [7].
Surface chemistry represents the defining frontier in quantum dot solids research, transitioning from a peripheral consideration to a central design parameter. The traditional view of ligands as passive spacers has evolved to encompass their roles as active mediators of charge transport, structural directors, and electronic modifiers. The correlation between ligand properties (length, functionality, binding group) and charge transport parameters (mobility, recombination, conductivity) provides a rational foundation for materials design.
Future developments will likely explore increasingly sophisticated ligand architectures including multi-functional systems, stimuli-responsive ligands, and hierarchically structured shells that address multiple challenges simultaneously. The integration of computational screening with high-throughput experimental synthesis will accelerate the discovery of optimal ligand chemistries for specific applications. As these advances mature, quantum dot solids with engineered surface chemistry will enable new generations of efficient, solution-processable electronic and energy conversion devices that leverage the unique properties of nanoscale materials.
The pursuit of high-performance quantum dot (QD) solids for optoelectronic devices represents a central theme in modern materials science. These solids, which are dense films of semiconductor nanocrystals, form the active layer in a new generation of solar cells, photodetectors, light-emitting diodes, and memory devices [9] [10]. A critical and often determining factor in their performance is the efficiency of charge transport between individual quantum dots. This transport is governed not by the intrinsic properties of the nanocrystals alone, but predominantly by the molecular-scale environment surrounding them—specifically, the organic surface ligands [11].
Surface ligands are molecular chains that passivate the QD surface, preventing aggregation and ensuring colloidal stability during synthesis. However, when QDs are assembled into a solid film, these same ligands become the medium through which charges must travel to hop from one dot to the next. Long-chain, aliphatic ligands—such as oleic acid (OA) and trioctylphosphine (TOP)—which are ubiquitous in colloidal synthesis, create a significant physical and electronic barrier to this process. They act as insulating spacers, forcing charge carriers to undergo quantum mechanical tunneling through a potential barrier, thereby rendering the QD solid an electrical insulator [11].
This article provides an in-depth technical examination of the insulator-conductor transition in quantum dot solids, focusing on the fundamental role of long-chain ligands in creating a tunneling barrier. We will explore the underlying mechanisms, quantify the impact of various ligand treatments, and detail the experimental methodologies that enable researchers to actively engineer charge transport properties, thereby transforming an insulating QD solid into a conductive functional material.
The Fundamental Mechanism: Ligands as Tunneling Barriers
The electronic structure of a single quantum dot is characterized by discrete, atom-like energy levels due to quantum confinement. When QDs are brought into close proximity to form a solid, their wavefunctions can overlap, allowing for the possibility of band-like transport. However, this ideal scenario is disrupted by the presence of surface ligands.
The Tunneling Barrier Model
Long-chain aliphatic ligands, typically with 8 to 18 carbon atoms, function as passive spacers between the conductive inorganic cores of the QDs [4] [11]. The charge transport mechanism in such a system is dominated by thermally activated hopping. The rate of electron hopping between two adjacent QDs is exponentially dependent on the center-to-center distance, which is the sum of the QD diameter and the ligand barrier length.
The tunneling probability through such a barrier can be described by the simplified relationship: [ T \propto e^{-\beta d} ] where ( T ) is the tunneling probability, ( d ) is the tunneling distance (dictated by the ligand chain length), and ( \beta ) is a decay constant that depends on the height of the potential barrier. The potential barrier height is directly related to the energy difference between the conductive states of the QD and the molecular orbitals of the ligand. For saturated hydrocarbons, this barrier is large, making them excellent insulators [11]. Consequently, the macroscopic conductivity ( \sigma ) of the QD solid follows a similar exponential decay: [ \sigma \propto e^{-\beta d} ] This exponential dependence means that even small reductions in the inter-dot spacing can lead to dramatic, orders-of-magnitude increases in film conductivity.
Consequences for Device Performance
The insulating nature of long-chain ligands has profound implications for QD-based devices. In photovoltaics, low conductivity leads to high series resistance, reducing the fill factor and overall power conversion efficiency. The charges photogenerated within the film must travel through a network of QDs interconnected by these tunneling barriers to be collected at the electrodes. If the hopping probability is too low, carriers recombine before they can be extracted, resulting in lost photocurrent [12]. Similarly, in memory devices and transistors, low carrier mobility impedes switching speeds and overall device performance [10]. Therefore, overcoming the insulating barrier presented by native long-chain ligands is a critical first step in realizing functional QD electronic devices.
Quantitative Evidence: Measuring the Ligand Impact
The influence of surface ligands on the electronic properties of QD solids is not merely qualitative; it can be directly measured and quantified through structural and optoelectronic characterization.
Ligand-Dependent Inter-Dot Spacing
Direct evidence of the spacer function of ligands comes from transmission electron microscopy (TEM). Studies on PbS QD films have precisely measured the reduction in interparticle distance following exchanges of long ligands for shorter ones.
Table 1: Impact of Ligand Treatments on Inter-Dot Spacing in PbS QD Films
| Ligand Treatment | Backbone Description | Average Inter-Dot Distance (nm) | Change vs. Oleic Acid |
|---|---|---|---|
| Oleic Acid (OA) | Long aliphatic (C18) | 10.2 ± 0.8 | Reference |
| 1,3-Benzenedithiol (1,3-BDT) | Conjugated aromatic | 9.3 ± 0.8 | -0.9 nm |
| 1,2-Ethanedithiol (EDT) | Short aliphatic (C2) | 7.8 ± 0.8 | -2.4 nm |
| Mercaptopropionic Acid (MPA) | Short aliphatic (C3) | 7.6 ± 0.8 | -2.6 nm |
| Ammonium Sulfide ((NH₄)₂S) | Atomic sulfide | 6.7 ± 0.8 | -3.5 nm |
Data adapted from [11].
The data in Table 1 clearly demonstrates that ligand exchange directly modulates the physical separation between QDs. The replacement of oleic acid with shorter molecules like EDT and MPA reduces the inter-dot distance by approximately 25%, significantly lowering the tunneling barrier. The minimal spacing achieved with atomic ligands like sulfide illustrates the ultimate limit of this approach, bringing the QDs into near-contact [11].
Correlation with Optoelectronic Properties
The structural changes induced by ligand exchange have a direct and measurable impact on the optoelectronic properties of the film. Time-resolved photoluminescence (TRPL) measurements reveal how ligand treatments alter charge carrier dynamics.
Films with long, insulating ligands exhibit longer photoluminescence (PL) lifetimes. This is because the excited electron-hole pair (exciton) is spatially confined to a single dot, as the probability of tunneling apart is low. When shorter, conductive ligands are introduced, the PL lifetime typically decreases dramatically. This PL quenching is a signature of enhanced charge transfer between QDs; the exciton can now dissociate, with the electron and hole hopping to neighboring dots, where they may encounter non-radiative recombination centers or be extracted as current [11].
Furthermore, the performance of working devices validates this trend. Solar cells based on PbS QDs consistently show that exchanging oleic acid for shorter thiols like EDT or MPA leads to a substantial increase in photocurrent and overall efficiency, a direct consequence of improved charge carrier collection [12] [13].
Beyond Passive Spacers: Active and Hybrid Ligand Systems
The prevailing strategy to overcome the tunneling barrier has been to shorten the ligand or replace it with an inorganic atom. However, a paradigm shift is emerging, moving from viewing ligands as passive spacers to using them as active components in the charge transport process.
Redox-Active Ligands
A groundbreaking approach involves the use of redox-active ligands, which introduce distinct electronic states within the bandgap that can actively mediate charge transport. As demonstrated in ZnO quantum dot systems, ligands like ferrocene carboxylate (FcCOO⁻) provide an alternative pathway for charge movement [4].
In this model, illustrated in the diagram below, charge transport occurs via two complementary mechanisms:
- Direct Hopping: Electron transfer through the conduction band of the QDs.
- Self-Exchange: Charge transfer via the immobilized redox ligands, where a charge on one ligand site hops to an adjacent, unoccupied ligand site through a chain reaction.
This dual-pathway model was experimentally confirmed using an electrochemical gating technique, which showed that the rate of the self-exchange process could be rationally controlled by modulating the Fermi level and the surface coverage of the redox ligands [4]. This represents a shift from passive barrier reduction to active transport pathway engineering.
The Mobility-Invariant Regime and Trap Limitation
A critical insight from recent research is that simply improving the theoretical mobility by shortening ligands is not always sufficient. In many practical CQD films, the diffusion length (LD) of charge carriers is not limited by mobility but by the presence of a low density of severe trap states [12].
In this trap-limited regime, the diffusion length is governed by the average distance a carrier can travel before encountering a trap, not by how fast it can move. The relationship is given by ( LD \approx d \times \sqrt{N{hops}} ), where ( d ) is the dot-to-dot distance and ( N_{hops} ) is the number of hops a carrier can make before being trapped [12]. In this scenario, increasing the mobility (e.g., by shortening ligands) only brings the carrier to the trap faster, without extending the diffusion length. This explains why some high-mobility CQD solids do not yield high-efficiency photovoltaic devices. The focus, therefore, must expand to include defect passivation in addition to inter-dot coupling.
Experimental Protocols for Ligand Exchange and Characterization
Engineering the insulator-conductor transition requires reliable and reproducible experimental protocols. The following sections detail standard methodologies for ligand exchange and subsequent characterization.
Solid-State Ligand Exchange
This is the most common method for fabricating conductive QD films and involves a two-step process: film deposition with native ligands, followed by in-situ ligand exchange.
Detailed Protocol:
- Substrate Preparation: Clean the substrate (e.g., ITO-glass, SiO₂/Si) with oxygen plasma or UV-ozone treatment to ensure a hydrophilic, clean surface.
- QD Film Deposition: Spin-coat a concentrated solution of QDs (capped with oleic acid or similar long ligands) in a non-polar solvent (e.g., toluene, octane) onto the substrate. Multiple layers may be spun to achieve the desired thickness, with mild annealing (e.g., 70°C for 5-10 minutes) between layers to remove solvent.
- Ligand Exchange Immersion: Immerse the film in a 0.01 - 0.1 M solution of the new, short ligand in a polar solvent (e.g., acetonitrile for EDT, methanol for MPA). The immersion time can vary from a few seconds to several minutes, depending on the ligand and film thickness.
- Rinsing and Drying: Immediately after immersion, rinse the film thoroughly with the same polar solvent (e.g., acetonitrile) to remove unbound ligand and reaction byproducts. Dry the film under a stream of nitrogen or with a brief anneal (e.g., 80-100°C for 1-5 minutes).
In-Solution Ligand Exchange
This method involves performing the ligand exchange on the QDs while they are still in solution, prior to film deposition. This can lead to more uniform ligand coverage and higher quality films.
Detailed Protocol:
- Precipitation and Redispersion: Precipitate the pristine QDs from their native solution by adding a non-solvent (e.g., ethanol or acetone). Centrifuge the mixture to obtain a pellet, and decant the supernatant.
- Ligand Exchange: Redisperse the QD pellet in a solution containing the new ligand (e.g., a chlorothiol in toluene). The concentration of the ligand is typically in large excess compared to the estimated number of surface sites on the QDs.
- Incubation: Stir or shake the mixture for a period ranging from hours to days to allow for complete ligand exchange.
- Purification: Precipitate and centrifuge the QDs again to remove the displaced native ligands and excess free ligands. Redisperse the final product in a suitable solvent for film deposition.
This method, particularly with robust ligands like chlorothiols, has been shown to produce films with lower trap densities, as the QDs are never exposed to a bare, unprotected surface during processing [12].
Essential Research Reagents and Materials
Table 2: The Scientist's Toolkit: Key Reagents for Ligand Engineering Studies
| Reagent/Material | Function/Description | Example in Context |
|---|---|---|
| Oleic Acid (OA) | Native long-chain (C18) insulating ligand; provides colloidal stability. | Standard ligand from synthesis; reference point for insulating behavior [11]. |
| 1,2-Ethanedithiol (EDT) | Short-chain (C2) dithiol ligand; significantly reduces inter-dot distance. | Common solid-state exchange ligand; induces conductivity in PbS films [11] [12]. |
| Mercaptopropionic Acid (MPA) | Short-chain (C3) ligand with thiol and carboxylic acid groups. | Used in PV devices; can bind in a bidentate fashion to PbS QDs [11] [12]. |
| Ferrocene Carboxylic Acid | Redox-active ligand; provides active charge transport states. | Enables self-exchange charge transport pathway in ZnO QD films [4]. |
| Ammonium Sulfide ((NH₄)₂S) | Inorganic atomic ligand; minimizes inter-dot spacing. | Creates inorganically interconnected QD solids with minimal tunneling barrier [11]. |
| Chlorothiols (e.g., 2-Chloroethanethiol) | In-solution passivation ligand; forms strong bond with QD surface. | Provides robust passivation from solution to film, reducing trap states [12]. |
| Acetonitrile & Methanol | Polar solvents for ligand exchange solutions and rinsing. | Used to process EDT and MPA solutions, respectively; do not dissolve the QD film. |
The transition of a quantum dot solid from an insulator to a conductor is a process meticulously engineered at the molecular level, primarily through the strategic manipulation of surface ligands. Long-chain, aliphatic ligands unequivocally create a formidable tunneling barrier that impedes charge transport, defining the insulating state. The primary strategy to overcome this has been ligand exchange with shorter molecules or atomic species, which reduces the inter-dot spacing and lowers the tunneling barrier.
However, contemporary research has revealed a more nuanced picture. The emergence of redox-active ligands introduces a paradigm where the ligand shell is not an obstacle to be minimized but an active medium for charge transport. Furthermore, the understanding of trap-limited transport highlights that maximizing mobility is insufficient without concurrently passivating mid-gap states that act as recombination centers. The future of high-performance QD solids lies in integrated optimization strategies that combine ligand engineering, interface engineering, and defect passivation to simultaneously enhance electronic coupling and maximize carrier lifetime. This holistic approach, potentially guided by machine learning for materials discovery, will be crucial for unlocking the full potential of quantum dot solids in next-generation optoelectronic devices.
The electronic and optical properties of semiconducting nanocrystals, or quantum dots (QDs), deviate significantly from their bulk counterparts due to quantum confinement. When the physical size of a semiconductor particle becomes comparable to or smaller than the Bohr exciton radius, the motion of charge carriers (electrons and holes) is spatially confined. This confinement quantizes the energy levels, leading to size-dependent properties, a phenomenon central to their application in optoelectronics and biomedicine. This whitepaper details the fundamental principles of quantum confinement and the Bohr exciton radius, framing them within the critical context of charge transport in quantum dot solids, where surface ligand engineering is a key determinant of device performance.
Core Principles: The Bohr Exciton Radius
The Bohr exciton radius ((a_B)) is the natural length scale for quantifying the onset of quantum confinement. It represents the average physical separation between the electron and hole in a bound electron-hole pair (an exciton) in the bulk semiconductor material.
The Bohr exciton radius is given by: [ aB = \frac{4\pi\epsilon \hbar^2}{\mu e^2} ] where (\epsilon) is the dielectric constant of the semiconductor, (\hbar) is the reduced Planck's constant, (\mu) is the reduced mass of the exciton ((\frac{1}{\mu} = \frac{1}{me^} + \frac{1}{m_h^})), and (e) is the electron charge.
When the radius ((R)) of the QD is less than (aB), strong confinement occurs, where both the electron and hole are independently confined. This results in a discretization of the energy states and a widening of the band gap ((Eg)) with decreasing particle size. The relationship is often approximated by the Brus equation for spherical QDs: [ E{QD} = Eg + \frac{\hbar^2 \pi^2}{2R^2} \left( \frac{1}{me^*} + \frac{1}{mh^*} \right) - \frac{1.8e^2}{4\pi\epsilon R} ] where the second term represents the quantum localization energy and the third term accounts for the Coulomb attraction.
Table 1: Bohr Exciton Radii and Confinement Regimes for Common Quantum Dot Materials
| Semiconductor Material | Bulk Band Gap (eV) | Bohr Exciton Radius, (a_B) (nm) | Strong Confinement Regime (R < (a_B)) |
|---|---|---|---|
| CdSe | 1.74 | ~5.6 | R < ~5.6 nm |
| PbS | 0.41 | ~18 | R < ~18 nm |
| CdTe | 1.49 | ~7.5 | R < ~7.5 nm |
| ZnO | 3.37 | ~2.0 | R < ~2.0 nm |
| Perovskite (MAPbI(_3)) | ~1.6 | ~2-5 | R < ~2-5 nm |
The Ligand-Transport Nexus in Quantum Dot Solids
In a QD solid film, individual dots are passivated by organic surface ligands. The length and chemical nature of these ligands are not merely a synthetic detail; they are critical design parameters that directly influence inter-dot coupling and charge transport, which is the thesis context of this guide.
- Short Ligands: Promote closer inter-dot spacing, enhancing electronic coupling and wavefunction overlap. This leads to higher charge carrier mobility but can also increase the probability of Förster Resonance Energy Transfer (FRET), potentially competing with charge transport.
- Long Ligands: Act as thicker insulating barriers between QDs. While they provide excellent colloidal stability and passivation, they severely hinder charge transport by increasing the tunneling distance, thereby reducing mobility.
Table 2: Impact of Surface Ligand Chain Length on QD Solid Properties
| Ligand Type (Example) | Approx. Length | Inter-dot Spacing | Electronic Coupling | Charge Mobility | Stability & Processability |
|---|---|---|---|---|---|
| Oleic Acid (OA) | ~1.8 nm | Medium | Medium | Medium | High |
| Butylamine (BA) | ~0.7 nm | Small | High | High | Medium |
| Octadecylphosphonic Acid (ODPA) | ~2.3 nm | Large | Low | Low | Very High |
| Ligand Exchange (e.g., SCN⁻) | Atomic/Ionic | Minimal | Very High | Very High | Requires matrix support |
Experimental Protocols
Protocol: Synthesizing Size-Tuned CdSe Quantum Dots (Hot-Injection Method)
Objective: To produce a series of CdSe QDs with diameters from 2-6 nm to demonstrate quantum confinement.
Materials: Cadmium oxide (CdO), Selenium (Se) shot, Trioctylphosphine oxide (TOPO), Hexadecylamine (HDA), Trioctylphosphine (TOP), Oleic acid (OA), 1-Octadecene (ODE).
Procedure:
- Preparation: Load a mixture of CdO (0.0128 mmol), OA (0.2 mL), and ODE (5 mL) into a 25 mL three-neck flask. Heat to 150°C under argon until a clear solution forms.
- Selenium Stock: In a glove box, dissolve Se shot (0.1 mmol) in TOP (0.5 mL) and ODE (1.5 mL) to form a TOP-Se solution.
- Injection & Growth: Raise the temperature of the cadmium solution to 300°C under vigorous stirring. Rapidly inject the TOP-Se solution. The growth temperature and time control the final size.
- For 2 nm dots: Inject at 240°C, quench immediately.
- For 4 nm dots: Inject at 300°C, grow for 60 seconds.
- For 6 nm dots: Inject at 300°C, grow for 10 minutes.
- Purification: Cool the reaction flask to 60°C. Add toluene and precipitate the QDs with ethanol. Centrifuge and redisperse in a non-polar solvent (e.g., hexane).
Protocol: Ligand Exchange and Solid Film Fabrication
Objective: To create QD solid films with controlled inter-dot spacing using ligands of varying chain lengths.
Materials: As-synthesized QDs (e.g., CdSe-OA), Ligand solutions (e.g., Butylamine, Octadecylamine, 0.1 M Potassium Thiocyanate in methanol), Solvents (Methanol, Butanol, Toluene).
Procedure:
- Ligand Exchange: Precipitate 1 mL of the purified QD solution with methanol. Centrifuge and discard the supernatant.
- Redisperse the QD pellet in 2 mL of a solution containing the new ligand (e.g., 50 µL butylamine in 2 mL methanol). Vortex and shake for 1 hour.
- Precipitate the ligand-exchanged QDs with a non-solvent (e.g., butanol), centrifuge, and redisperse in an appropriate solvent (e.g., butanol for short ligands, toluene for long ligands).
- Film Fabrication: Deposit the QD solution onto a pre-cleaned substrate (e.g., glass/ITO) via spin-coating (e.g., 2000 rpm for 30 seconds) or drop-casting. Anneal the film at 80-100°C for 10 minutes to remove residual solvent.
Protocol: Characterizing Confinement and Charge Transport
Objective: To measure the optical properties and electrical performance of the QD films.
Materials: UV-Vis-NIR Spectrophotometer, Photoluminescence (PL) Spectrometer, Field-Effect Transistor (FET) test structure with pre-patterned electrodes, Semiconductor Parameter Analyzer.
Procedure:
- Optical Characterization:
- Dilute a sample of each QD size in toluene and record the UV-Vis absorption spectrum. The position of the first excitonic peak is used to determine the QD size using established calibration curves.
- Record the PL spectrum to determine the emission maximum and Full Width at Half Maximum (FWHM), which indicates size distribution.
- Structural Characterization: Use Transmission Electron Microscopy (TEM) to verify QD size, shape, and monodispersity.
- Charge Transport Measurement:
- Fabricate a bottom-gate, top-contact FET using the ligand-exchanged QD film as the channel.
- Using a semiconductor parameter analyzer, sweep the gate voltage ((VG)) at a fixed drain-source voltage ((V{DS})).
- Extract the field-effect mobility ((\mu{FET})) from the slope of the transfer curve ((I{DS}) vs (VG)) in the saturation regime using the standard MOSFET equation: (I{DS} = (\frac{W}{2L}) Ci \mu{FET} (VG - VT)^2), where (C_i) is the gate dielectric capacitance, and (W/L) is the channel width-to-length ratio.
Visualizations
Diagram 1: Quantum Confinement Effect (77 chars)
Diagram 2: Ligand Length Impact on Transport (75 chars)
Diagram 3: QD Solid Fabrication Workflow (73 chars)
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials for QD Synthesis and Fabrication
| Item | Function / Role |
|---|---|
| Cadmium Oxide (CdO) | Cadmium precursor for high-temperature synthesis of Cd-based QDs. |
| Trioctylphosphine Oxide (TOPO) | A common coordinating solvent and surfactant that controls QD growth and passivates the surface. |
| 1-Octadecene (ODE) | A non-coordinating, high-boiling-point solvent used in place of traditional coordinating solvents. |
| Oleic Acid (OA) | A common ligand that binds to the QD surface, providing colloidal stability and size control. |
| Trioctylphosphine (TOP) | A strong coordinating solvent used to prepare precursor stocks (e.g., TOP-Se, TOP-S). |
| Potassium Thiocyanate (KSCN) | A source of short, inorganic SCN⁻ ligands for ligand exchange to create strongly coupled QD solids. |
| Butylamine / Octadecylamine | Short- and long-chain amine ligands used to systematically study the effect of ligand length on transport. |
| Zinc Acetate / Zinc Oleate | Precursors for the fabrication of a ZnS or ZnO shell in core/shell QDs to enhance photoluminescence quantum yield. |
Colloidal nanocrystal quantum dots (QDs) assembled into solid-state films represent a promising class of semiconductors for next-generation, solution-processed electronic and optoelectronic devices. Their potential has been demonstrated in transistors, light-emitting diodes, solar cells, and photodetectors. Unlike bulk semiconductors, QD solids offer a multi-dimensional design space where electronic properties can be tuned by controlling quantum dot size, shape, composition, surface termination, and packing geometry [14]. Despite the commercial success of QDs as optical absorbers and emitters, applications relying on efficient charge transport have faced significant challenges due to the inability to predictively control their electronic properties [14]. The fundamental mechanism governing charge transport in these materials has remained elusive, hindering the development of predictive models for device design.
Central to understanding charge transport in QD solids is the recognition that charge carriers (electrons and holes) significantly interact with the atomic lattice of the nanocrystals and their surface environments. When a charge carrier localizes on an individual QD, it induces structural distortions through electrostatic interactions—primarily with the surface ligands. This coupled entity of charge carrier and lattice distortion forms what is known as a polaron [14]. The energy required to rearrange the atomic structure during charge transfer between neighboring QDs, termed the reorganization energy (λ), is substantial (tens to hundreds of meV) [14]. This polaron formation, combined with relatively weak electronic coupling between quantum dots, dictates that charge transport in typical QD solids does not occur through conventional band-like transport but rather through a phonon-assisted hopping process [14]. This whitepaper explores the physical principles of the polaron hopping model and examines how surface ligand engineering serves as a powerful tool to manipulate this charge transport mechanism.
The Physical Basis of Polaron Hopping
Polaron Formation and Reorganization Energy
The journey of a charge through a QD solid begins with its localization on an individual nanocrystal. Large-scale ab initio calculations on lead sulfide (PbS) QDs with iodine ligands have revealed that introducing an extra electron or hole induces measurable structural changes. Specifically, the Pb-ligand bonds on the (111) surfaces expand or contract, while the Pb-S bond lengths in the core remain largely unaffected [14]. Although these bond length changes are small (up to 0.2% of the nominal bond length), the collective reorganization energy λ is significant.
The reorganization energy λ decreases with increasing QD size due to two key factors: a reduced carrier density across the nanocrystal and an increased number of ligands sharing the structural distortion [14]. This size-dependent reorganization energy plays a critical role in determining charge transfer rates between quantum dots.
Table 1: Key Parameters in Polaron Hopping Transport
| Parameter | Symbol | Definition | Impact on Transport |
|---|---|---|---|
| Reorganization Energy | λ | Energy for structural rearrangement during charge transfer | Large λ (10s-100s meV) inhibits transfer; decreases with QD size |
| Electronic Coupling | Vct | Quantum mechanical overlap between neighboring QD states | Small Vct compared to λ leads to localized polarons |
| Transfer Rate | kct | Speed of charge hopping between QDs | Dictated by Marcus non-adiabatic model; peaks when ΔE ≈ -λ |
Electronic Coupling Between Quantum Dots
The electronic coupling, Vct, between neighboring QDs determines the degree of wavefunction overlap and thus the probability of charge transfer. In assembled PbS QD superlattices, the coupling strength depends strongly on the relative orientation of adjacent nanocrystals. Calculations show that coupling in the [100] direction is approximately an order of magnitude larger than in the [111] direction, as quantum confinement strongly localizes charge carriers away from the ligand-rich [111] facets [14].
Crucially, across a wide range of QD sizes and inter-dot spacings, Vct remains more than an order of magnitude smaller than the reorganization energy λ [14]. This relationship confirms that charge carriers exist as polarons localized on individual QDs and that transport occurs through phonon-assisted hopping between these localized states rather than through delocalized band transport.
The Phonon-Assisted Charge Transfer Mechanism
The significant disparity between Vct and λ places polaron hopping firmly in the non-adiabatic regime described by Marcus electron transfer theory. In this framework, charge transfer requires thermal activation to overcome the reorganization energy barrier. The atomic vibrations (phonons) of the Pb-ligand bonds, which occur at energies below ~15 meV, provide the necessary thermal energy to drive this process [14].
At temperatures above approximately 175 K, the charge transfer rate kct follows a Marcus-type expression [14]:
Where NP represents the number of degenerate product states, ΔE is the energy difference between initial and final states (including contributions from an applied electric field or energetic disorder), kB is Boltzmann's constant, and T is temperature. This model predicts room-temperature charge transfer times on the order of 10-100 picoseconds for PbS QD solids, consistent with experimental measurements [14].
Surface Ligands as a Control Parameter for Charge Transport
Ligand Engineering Fundamentals
Surface ligands play a dual role in QD solids: they passivate surface states to prevent charge trapping and mediate electronic coupling between neighboring nanocrystals. The nature of the ligand-QD bond and the physical properties of the ligand itself directly influence the charge transport mechanism. Ligands are typically classified as X-type (anionic, e.g., halides, thiols, carboxylates) or L-type (Lewis bases, e.g., amines, phosphines) [14].
In PbS QD systems, the use of X-type ligands leads to polaron formation through electrostatic interaction of the charge carrier with the negatively charged functional groups of the ligands [14]. The structural reorganization associated with polaron formation primarily occurs in the metal-ligand bonds, making the ligand identity and bonding geometry crucial determinants of the reorganization energy λ.
Ligand Chain Length and Electronic Coupling
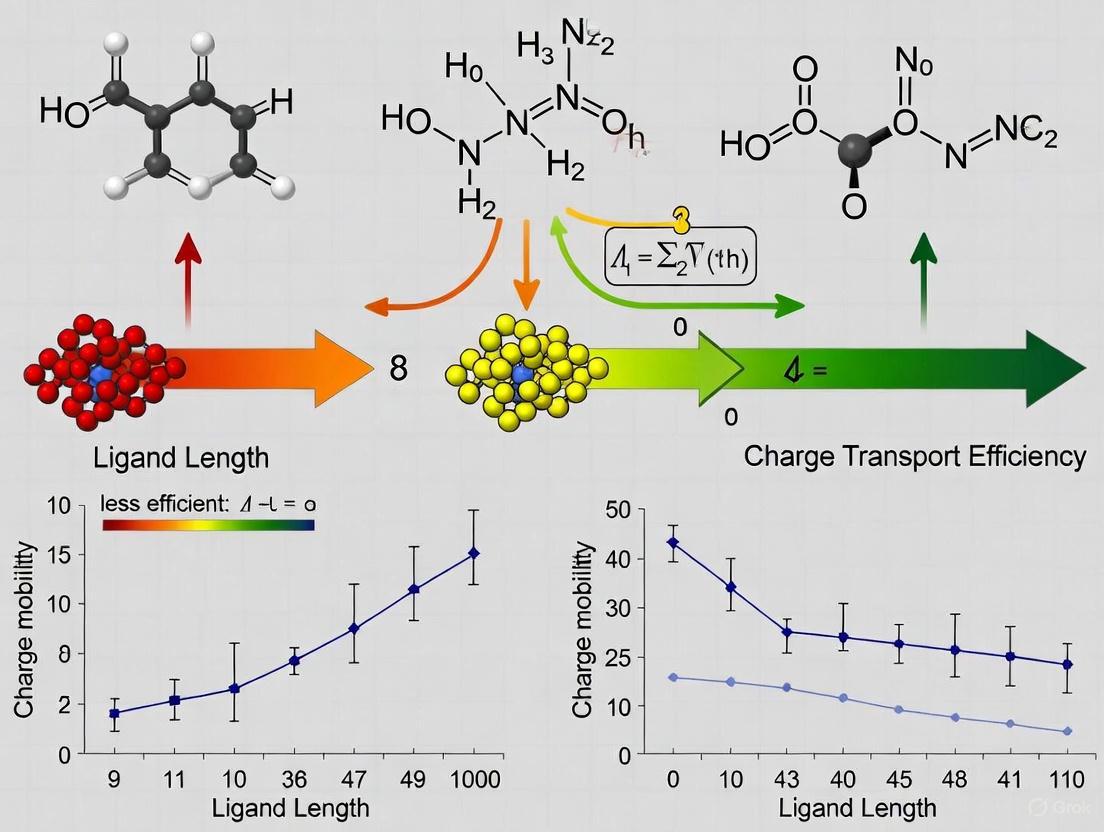
The length of the surface ligand alkyl chain directly impacts the electronic coupling Vct between quantum dots by determining the inter-dot separation. Studies on InP/ZnSe/ZnS QDs with systematically varied ligand chain lengths (oleic acid [OA], decanoic acid [DA], and hexanoic acid [HA]) demonstrate that shorter ligands significantly enhance charge injection efficiency [15]. Chronoamperometry measurements reveal that decreasing the carbon atom count in the capping ligand from 18 (OA) to 6 (HA) increases the integrated current density by a factor of 1.5 [15]. This enhancement is attributed to the reduced energy barrier for charge transport with shorter ligand chains.
Table 2: Ligand-Dependent Charge Transport Properties
| QD Material | Ligand Type | Chain Length/Type | Transport Property | Effect vs Long Chain |
|---|---|---|---|---|
| InP/ZnSe/ZnS | Carboxylic acids | C18 vs C6 | Current density | 1.5× increase [15] |
| PbS | n-butylamine | ~0.6 nm | Conductivity | 180× vs. drop-cast films [16] |
| ZnO | Ferrocene carboxylate | Redox active | Transport mode | Adds self-exchange pathway [4] |
| PbS | Iodine | X-type inorganic | Reorganization energy | Polaronic transport [14] |
Furthermore, ligand chain length affects inter-QD energy transfer dynamics, which competes with charge transport in optoelectronic devices. Fluorescence lifetime imaging microscopy (FLIM) of InP/ZnSe/ZnS QD solids reveals that energy transfer population and efficiency between neighboring QDs are proportional to surface ligand chain lengths [15]. X-ray diffraction analysis confirms that shorter alkyl ligands result in weaker interligand van der Waals interactions, reducing Förster resonance energy transfer (FRET) and potentially favoring charge transport over energy transfer [15].
Advanced Ligand Strategies
Beyond simple alkyl chains, researchers have developed sophisticated ligand engineering strategies to enhance charge transport in QD solids:
Hybrid Ligand Exchange: Combining solution-based exchange with S²⁻ and solid-state exchange with N³⁻ for InP QDs enhances interdot coupling and controls n-doping, resulting in electron mobilities of 0.45 cm² V⁻¹ s⁻¹—approximately 10 times higher than previously reported devices [6].
Redox-Active Ligands: Incorporating ferrocene carboxylate ligands on ZnO QDs introduces an alternative charge transport pathway through self-exchange reactions between immobilized redox centers [4]. This approach represents a shift from using ligands as passive spacers to employing them as active electronic components that directly participate in charge transport.
Inorganic Ligands: Replacing organic ligands with shorter inorganic ligands (e.g., halides, chalcogenides) significantly reduces inter-dot spacing and enhances electronic coupling. Surface modification of InP QDs with thin Se overlayers lowers trap-state densities and extends carrier diffusion lengths [6].
Experimental Evidence and Characterization Techniques
Nano-patterning for Reduced Disorder
Understanding the intrinsic charge transport properties of QD solids requires minimizing structural disorder that often obscures the fundamental mechanisms. Nano-patterning techniques have been developed to fabricate QD solids free of cracking, clustering, and grain boundaries [16]. These structurally optimized arrays exhibit conductivities 180 times higher than drop-cast films of the same QD material [16].
In narrow (70 nm) nano-patterned PbS QD solids with n-butylamine ligands, researchers have observed exceptionally large conductance noise exceeding 100% of the average current [16]. This noise displays a power-law spectral density rather than characteristic 1/f behavior, indicating complex dynamics of charge trapping and release rather than simple shot noise. The noise magnitude grows with increasing conductance under applied bias, gate voltage, and temperature, suggesting that the transport occurs through stochastic quasi-one-dimensional percolation paths [16].
Spectroscopic and Electrochemical Characterization
Multiple experimental techniques provide insights into the polaron hopping mechanism and ligand effects:
Chronoamperometry: This electrochemical technique monitors current from Faradaic processes at electrodes over time, revealing that shorter ligand chains significantly enhance charge injection efficiency into InP/ZnSe/ZnS QDs [15].
Fluorescence Lifetime Imaging Microscopy (FLIM): By mapping spatial variations in photoluminescence lifetimes, FLIM quantifies inter-QD energy transfer efficiency, which competes with charge transport and is strongly influenced by ligand chain length [15].
Single-QD Electrophoresis: Advanced laser scanning microscopy combined with high-field electrophoresis enables measurement of elementary charges on individual QDs in their native liquid environment, providing unprecedented insight into charging heterogeneity at the single-particle level [17].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for QD Charge Transport Studies
| Reagent Category | Specific Examples | Function & Rationale |
|---|---|---|
| QD Core Materials | PbS, InP, ZnO, CdSe/CdS | Base semiconductor determining band structure and quantum confinement [14] [4] [17] |
| Short Alkyl Ligands | n-butylamine, hexanoic acid | Reduce inter-dot distance, enhance electronic coupling V_ct [16] [15] |
| Inorganic Ligands | Iodine, sulfide, selenide | Maximize electronic coupling through minimal spacing and covalent bonding [14] [6] |
| Redox-Active Ligands | Ferrocene carboxylate | Introduce alternative charge transport via self-exchange reactions [4] |
| Solvents | Acetonitrile, dichloromethane, dodecane | Medium for ligand exchange and assembly; affects film morphology [4] [17] |
| Stabilizers | Polyisobutylene succinic anhydride | Maintain colloidal stability during single-QD measurements [17] |
The polaron hopping model provides a comprehensive framework for understanding charge transport in quantum dot solids as a phonon-assisted process. The critical relationship Vct << λ establishes that charge carriers form polarons localized on individual QDs, requiring thermal activation for inter-dot transfer. Surface ligand engineering emerges as a powerful strategy for controlling this transport mechanism, primarily through tuning both the reorganization energy λ (via the ligand-QD bond properties) and the electronic coupling Vct (via inter-dot spacing and chemical functionality).
Future research directions will likely focus on sophisticated ligand design that goes beyond simple spacing functions. Redox-active ligands that provide alternative charge transport pathways [4], hybrid exchange strategies that enhance coupling while minimizing disorder [6], and precise nano-patterning techniques that eliminate structural defects [16] represent promising approaches to achieving more predictable and efficient charge transport in QD solids. A fundamental understanding of the polaron hopping mechanism and its dependence on surface chemistry will accelerate the development of next-generation QD-based electronic devices with tailored performance characteristics.
Soft and Hard Acid-Base (HSAB) Theory as a Guide for Ligand Selection and Exchange
The Hard and Soft Acids and Bases (HSAB) theory, introduced by Ralph Pearson, provides a powerful conceptual framework for predicting the stability and reactivity of Lewis acid-base pairs [18]. This principle classifies chemical species based on their polarizability, not their strength, dividing them into "hard" or "soft" categories [19]. In quantum dot (QD) science, this translates to a critical design rule: metal centers (acids) interact most strongly and form more stable complexes with ligands (bases) of matching character—hard with hard, soft with soft [18] [20]. This selective affinity is the cornerstone of rational ligand design for controlling surface chemistry in quantum dot solids, a factor that directly governs their charge transport properties [1] [21].
The surface of a QD is a dynamic interface where the inorganic core meets its organic ligand environment. The nature of this interface dictates the electronic coupling between neighboring dots in a solid film, influencing everything from carrier mobility to environmental stability [3] [1]. Applying HSAB theory allows researchers to move beyond trial-and-error approaches. By rationally selecting ligands based on the hardness or softness of the surface metal atoms (e.g., Pb²⁺ in PbS QDs), one can predict bonding stability, anticipate the kinetics of ligand exchange, and ultimately tailor materials for enhanced performance in devices like photovoltaics and photodetectors [2] [21].
Fundamental Principles of HSAB Theory
The classification of a species as "hard" or "soft" depends on its intrinsic electronic properties, primarily its polarizability—that is, the ease with which its electron cloud can be distorted [19] [18].
Defining Characteristics
Hard acids and bases are typically small in ionic radius, have high charge states, and exhibit low polarizability. They form bonds that are predominantly ionic in nature [19] [18]. Their electron density is tightly held, making them less susceptible to deformation.
Soft acids and bases, in contrast, are generally larger, have lower charge states, and are highly polarizable. They tend to form bonds with significant covalent character [19] [18]. Their diffuse electron clouds are easily distorted, facilitating orbital overlap that leads to covalent bonding.
Table 1: Key Properties of Hard and Soft Acids and Bases
| Property | Hard Acids/Bases | Soft Acids/Bases |
|---|---|---|
| Atomic/Ionic Radius | Small | Large |
| Charge Density | High | Low |
| Polarizability | Low | High |
| Preferred Bonding | Ionic | Covalent |
| Electronegativity (Bases) | High | Low |
Classification of Common Species in QD Chemistry
Understanding the classification of specific metal ions and ligands is essential for applying HSAB theory [18].
Hard Acids: Characteristic hard acids include small, highly charged metal ions such as H⁺, Li⁺, Na⁺, K⁺, and Al³⁺. In the context of QDs, early transition metal ions in high oxidation states also fall into this category [19].
Soft Acids: These include metal ions with a low charge density and high polarizability. Notably, many metals used in QDs are soft or borderline acids. For example, Pb²⁺ is classified as a borderline acid, while Cd²⁺, Hg²⁺, and Au⁺ are typical soft acids [18]. The bulk metal atoms (M⁰) at a QD's surface also exhibit soft character [18].
Hard Bases: These feature small donor atoms with high electronegativity, such as oxygen or fluorine. Common examples are hydroxide (OH⁻), fluoride (F⁻), carbonate (CO₃²⁻), and carboxylate groups (RCOO⁻) like acetate [19] [18]. Water and ammonia are also hard bases.
Soft Bases: Soft bases contain large, highly polarizable donor atoms like sulfur, phosphorus, or iodine. Key examples are thiolate (RS⁻), iodide (I⁻), thiocyanate (SCN⁻), and phosphines (PR₃) [19] [18].
Table 2: Common Acids and Bases in Quantum Dot Surface Chemistry
| Category | Examples |
|---|---|
| Hard Acids | H⁺, Na⁺, K⁺, Al³⁺ |
| Borderline Acids | Fe²⁺, Co²⁺, Pb²⁺, Zn²⁺ |
| Soft Acids | Cd²⁺, Hg²⁺, Ag⁺, Au⁺, Pt²⁺, Pd²⁺ |
| Hard Bases | H₂O, NH₃, OH⁻, F⁻, CH₃COO⁻, R-OH |
| Soft Bases | I⁻, R-S⁻ (Thiolates), SCN⁻, CO, PR₃ |
HSAB Theory in Quantum Dot Surface Chemistry
The surface of a quantum dot is composed of under-coordinated metal and chalcogenide ions, which act as Lewis acid and base sites, respectively. For lead chalcogenide QDs like PbS, the Pb²⁺ sites are borderline Lewis acids, while the S²⁻ sites are soft Lewis bases [18] [21]. This fundamental characteristic dictates the optimal ligand chemistry for effective passivation and property control.
Ligand Selection for Surface Passivation
Effective surface passivation requires satisfying the bonding preferences of both ionic constituents. According to HSAB theory, the borderline acidic Pb²⁺ sites show a strong thermodynamic preference for bonding with soft bases [18]. This explains the widespread effectiveness of soft thiolate-based ligands (R-S⁻) and iodide (I⁻) for passivating PbS QD surfaces [21]. These soft ligands form stable, covalent-like bonds with the lead atoms, reducing surface trap states and enhancing photoluminescence.
Conversely, the soft basic S²⁻ sites can be passivated by borderline or soft acids. In a practice known as Z-type passivation, metal halides like PbI₂ or CdI₂ are used, where the soft Pb²⁺ or Cd²⁺ ions bind to the sulfur sites [1] [21]. This co-passivation strategy—using a soft base for the metal sites and a soft acid for the chalcogen sites—creates a more robustly passivated and electronically stable QD surface.
Impact of Ligand Character on Charge Transport
The HSAB-driven selection of ligands directly impacts charge transport in QD solids through two primary mechanisms: trap state passivation and inter-dot coupling.
Trap State Passivation: An improperly passivated surface contains "dangling bonds" that act as electronic trap states, capturing charge carriers and impeding transport. Hard-soft mismatches (e.g., a hard base on a soft acid site) result in weak, labile bonds that readily desorb, creating traps. Using HSAB-predicted, complementary ligands ensures strong, stable bonding, which minimizes trap state density and reduces charge carrier recombination [3] [21]. This leads to longer carrier diffusion lengths, which is critical for high-performance devices like solar cells [2] [22].
Inter-Dot Coupling: The ligand shell acts as a physical and electronic barrier between QDs. Short, compact ligands chosen via HSAB principles allow for closer packing of QDs. This decreases the tunneling barrier width and enhances the electronic coupling between adjacent dots, facilitating band-like transport or more efficient hopping conduction [1]. For instance, replacing long, insulating oleic acid (a relatively harder base) with compact iodide (a soft base) on PbS QDs dramatically improves electron mobility in the solid state [1] [21].
Experimental Protocols for Ligand Exchange
Ligand exchange is a critical process for replacing long-chain, insulating native ligands with shorter, charge-transport-enhancing ligands. The following protocols, grounded in HSAB principles, are standard in the field.
Solid-State Ligand Exchange Using Metal Iodides
This protocol describes the conversion of oleic acid-capped PbS QDs to iodide-capped QDs for high-mobility solids [2] [21].
Principle: The soft character of the I⁻ ion provides a strong thermodynamic driving force to displace the harder carboxylate group (oleate) from the borderline acidic Pb²⁺ surface sites.
Materials:
- PbS-OA QDs: Oleic acid-capped PbS quantum dots in toluene (e.g., 50 mg/mL).
- Lead Iodide (PbI₂): Serves as the source of I⁻ ligands and Z-type passivant.
- Dimethylformamide (DMF) or Butylamine: Polar solvent to dissolve PbI₂ and facilitate exchange.
- Toluene, Methanol, Acetone: Solvents for washing and precipitation.
Procedure:
- Solution Preparation: Dissolve 100 mg of PbI₂ in 5 mL of DMF or butylamine under vigorous stirring and mild heating (~50°C) until fully dissolved.
- Film Deposition: Spin-coat a thin film of PbS-OA QDs onto the desired substrate (e.g., glass/ITO).
- Exchange Reaction: Gently drop-cast the PbI₂ solution onto the QD film and allow it to stand for 30-60 seconds. The film color will change, indicating the exchange.
- Washing: Rinse the film thoroughly with pure DMF to remove excess PbI₂ and displaced oleic acid, followed by a rinse with methanol or acetone.
- Drying: Dry the film under a stream of nitrogen or with a brief anneal on a hotplate (~70°C).
Notes: The concentration of PbI₂, solvent choice, and reaction time are key optimization parameters. Butylamine can act as a co-ligand and may accelerate the exchange process.
Solution-Phase Ligand Exchange with Short-Chain Organic Ligands
This protocol replaces native ligands with short, conductive organic molecules in solution, prior to film deposition [3] [21].
Principle: A large excess of the incoming ligand (e.g., a soft thiol) shifts the equilibrium to favor displacement of the native ligand, forming a QD ink ready for processing.
Materials:
- PbS-OA QDs: In toluene or hexane.
- Incoming Ligand: e.g., 1,2-ethanedithiol (EDT), 3-mercaptopropionic acid (MPA), or tetrabutylammonium iodide (TBAI).
- Solvents: Acetonitrile, methanol, ethanol, ethyl acetate (as non-solvents for precipitation).
Procedure:
- Precipitation: Add a non-solvent (e.g., acetone or methanol) to the pristine QD solution to precipitate the QDs. Centrifuge and discard the supernatant.
- Redispersion: Redisperse the QD pellet in a minimal amount of a solvent that is compatible with both the QDs and the incoming ligand (e.g., octane for EDT exchange).
- Ligand Addition: Add a large excess (e.g., 100-1000x molar relative to QDs) of the incoming ligand to the QD solution.
- Reaction: Stir the mixture for 1-24 hours at room temperature or with mild heating.
- Purification: Precipitate the ligand-exchanged QDs by adding a non-solvent, centrifuge, and discard the supernatant containing the displaced ligands.
- Final Ink: Redisperse the purified QD pellet in an appropriate solvent for film deposition (e.g., octane for EDT-capped QDs).
Notes: This method offers excellent control over the QD-to-ligand ratio. The choice of solvent for the final ink is crucial for achieving high-quality, crack-free films during deposition.
The Scientist's Toolkit: Essential Reagents and Materials
Successful ligand engineering requires a set of well-defined reagents and materials. The following table details key components for a typical HSAB-guided research workflow in QD science.
Table 3: Research Reagent Solutions for QD Ligand Engineering
| Reagent/Material | Function & HSAB Role | Example Application |
|---|---|---|
| Oleic Acid (OA) | Native surfactant (hard base); provides colloidal stability after synthesis. | Standard initial capping ligand for PbS, CdSe QD synthesis [21]. |
| Lead Iodide (PbI₂) | Source of I⁻ (soft base) and Pb²⁺ for Z-type passivation. | Solid-state ligand exchange on PbS QD films for photovoltaics [2] [21]. |
| 1,2-Ethanedithiol (EDT) | Short-chain dithiol ligand (soft base). | Solution-phase exchange to create conductive PbS QD solids for FETs [21]. |
| Tetrabutylammonium Iodide (TBAI) | Source of "naked" I⁻ ions (soft base) in solution. | Halide anion exchange for QDs, improving electron transport [21]. |
| 3-Mercaptopropionic Acid (MPA) | Short bifunctional ligand; thiol (soft base) binds QD, carboxylate aids solubility. | Creating water-soluble QDs or tuning surface polarity [21]. |
| Dimethylformamide (DMF) | Polar aprotic solvent (borderline base); dissolves metal halides. | Solvent for PbI₂ during solid-state ligand exchange [21]. |
| Butylamine | Polar solvent and L-type ligand (hard base); can coordinate to metal sites. | Accelerates ligand exchange processes [21]. |
Quantitative Data and Case Studies
The application of HSAB theory yields quantifiable improvements in QD device performance. Advanced spectroscopic techniques confirm the fundamental mechanisms at play.
Charge Transfer Dynamics
A 2025 study used Resonant Auger Spectroscopy (RAS) to probe ultrafast charge transfer dynamics in PbS QDs of different sizes and with different surface treatments [2]. The technique uses the core-hole lifetime as an internal clock to measure the rate at which an excited electron delocalizes from a Pb atom. The findings confirmed that larger QDs and those with optimized ligand environments (e.g., treated with PbI₂) exhibited faster charge transfer rates compared to smaller dots or poorly passivated references [2]. This directly links appropriate surface chemistry, guided by HSAB principles, to superior charge transport properties on the most fundamental timescales.
Photovoltaic Device Performance
The impact of HSAB-driven ligand selection is clearly demonstrated in the performance metrics of quantum dot solar cells (QDSCs). Devices utilizing inorganic ligands consistently outperform those with long-chain organic ligands.
Table 4: Impact of Ligand Choice on PbS Quantum Dot Solar Cell Performance
| Ligand Type | HSAB Classification | Typical Power Conversion Efficiency (PCE) | Key Effect on Transport |
|---|---|---|---|
| Oleic Acid (OA) | Hard Base | <5% | High inter-dot resistance, insulating film [21]. |
| Lead Iodide (PbI₂) | Soft Base / Z-type | >10% | Excellent passivation, high electron mobility [2] [21]. |
| 1,2-Ethanedithiol (EDT) | Soft Base | ~8-10% | Good hole conduction, short ligand length [21]. |
| Combined Halide/Organic | Mixed | >11% (Record devices) | Balanced charge transport, superior passivation [21] [22]. |
HSAB theory provides an indispensable, predictive framework for navigating the complex landscape of quantum dot surface chemistry. By guiding the rational selection of ligands based on the hard/soft character of the QD surface atoms, researchers can directly control critical outcomes: bonding stability, trap state density, and electronic coupling. The experimental protocols and quantitative data summarized herein demonstrate that moving from hard-hard or hard-soft mismatches to thermodynamically favored soft-soft pairs—such as employing iodide or thiolates for PbS QDs—is a decisive strategy for achieving high-performance electronic and optoelectronic devices. As QD research progresses toward more complex architectures and integration, the principles of HSAB will remain a cornerstone for the rational design of advanced functional materials.
Ligand Engineering in Action: Strategies for Enhanced Charge Transport
In quantum dot (QD) solids, the organic ligand shell surrounding the inorganic core is not merely a passive stabilizer but a critical component determining charge transport efficiency. These surface ligands dictate interparticle spacing, passivate surface states, and ultimately govern electronic coupling between adjacent QDs. Conventional ligand exchange strategies strategically replace long, insulating native ligands with shorter organic or inorganic counterparts to "shorten the bridge" between quantum dots, thereby enhancing charge carrier mobility while maintaining colloidal stability. This process is fundamental to developing high-performance QD-based electronic devices, including photodetectors, solar cells, and light-emitting diodes. The following sections provide a technical examination of ligand exchange methodologies, quantitative performance comparisons, and practical protocols for implementation.
Core Principles: How Ligand Engineering Modulates Charge Transport
The Interparticle Distance Mechanism
The most direct impact of ligand exchange is on the physical separation between adjacent quantum dots. Long-chain insulating ligands (e.g., oleic acid, oleylamine) create substantial tunnel barriers that impede charge carrier hopping. Replacing them with shorter ligands dramatically reduces the interparticle distance, leading to an exponential increase in the wavefunction overlap between neighboring QDs. For instance, replacing oleylamine (approximately 5.0 nm interparticle distance) with sulfide ligands on InSb QDs shortened the interparticle distance to 1.5 ± 0.5 nm, significantly enhancing carrier mobility [23]. This reduction directly decreases the tunneling barrier height and width, facilitating more efficient electron hopping between quantum dots.
Electronic Coupling and Trap State Passivation
Beyond mere physical proximity, the chemical nature of the replacement ligands determines their ability to mediate electronic coupling and passivate surface states. Different ligand classes exhibit distinct functionalities:
- Inorganic Ligands: Materials like sulfide (S²⁻) or halides (Br⁻, I⁻) provide atomic-scale bridging and effective passivation of anionic surface sites. On InSb QDs, sulfide capping yielded better carrier mobility and lower trap density compared to bromine capping [23].
- Short-Chain Organic Ligands: Molecules like butylamine (BA) and ethanedithiol (EDT) reduce distance while maintaining some organic character for processing.
- Conjugated Organic Ligands: Ligands with π-conjugated systems (e.g., oligo-(phenylene vinylene)) can actively mediate charge transport through electronic delocalization [24].
- Redox-Active Ligands: Molecular species like ferrocene carboxylate introduce electronic states that provide an additional pathway for charge transport via self-exchange reactions [4].
Proper ligand selection must balance multiple factors: achieving close-packed QD solids, maintaining sufficient surface passivation to reduce trap-assisted recombination, and providing favorable energy level alignment for specific device applications.
Quantitative Performance Comparison of Ligand Systems
Table 1: Comparative Performance Metrics of Different Ligand Classes in QD Solids
| Ligand Type | Specific Example | Interparticle Distance | Carrier Mobility Enhancement | Key Performance Metrics | Application Demonstrated |
|---|---|---|---|---|---|
| Inorganic | Sulfide (S²⁻) | 1.5 ± 0.5 nm (from 5.0 ± 0.5 nm) | Precedence over Br⁻ capping | EQE: 18.5%; Low dark current (~nA cm⁻²) | InSb SWIR photodiodes [23] |
| Short Organic | Ethanedithiol (EDT) | Significant reduction | ~90% ligand exchange in ≤60 s | Rapid processing; Compatible with printing | PbS QD microstructures [25] |
| Redox Active | Ferrocene carboxylate | Not specified | Enables self-exchange transport | Two complementary charge transport pathways | ZnO QD assemblies [4] |
| Short Organic | Butylamine (BA) | Intermediate reduction | Improved morphology preservation | Prevents void defects in polymer:QD blends | PTB1:Pbs solar cells [26] |
Table 2: Impact of Ligand Exchange on Photovoltaic Device Parameters
| Ligand System | QD Material | Dark Current Density | External Quantum Efficiency (EQE) | Response Speed | Stability Observations |
|---|---|---|---|---|---|
| Sulfide-capped | InSb | ~nA cm⁻² at 0 V | 18.5% | Enhanced (vs. bromide-capped) | Not specified [23] |
| Bromide-capped | InSb | Higher than sulfide | Lower than sulfide | Slower (second-scale) | Not specified [23] |
| OA to MPA direct | PTB1:PbS | Not specified | Not specified | Severe film morphology changes | Non-ideal morphology [26] |
| OA to BA to MPA | PTB1:PbS | Not specified | Not specified | Preserved morphology | Maintained BHJ structure [26] |
Experimental Protocols: Methodologies for Ligand Exchange
Solid-State Ligand Exchange (SSLE) for PbS QD Microstructures
This protocol enables efficient ligand exchange on pre-deposited QD films, particularly suitable for patterned structures [25]:
- QD Film Preparation: Fabricate PbS QD microstructures via electrohydrodynamic (EHD) printing using tetradecane as solvent for optimal spatial resolution. Achieve varying thicknesses (125-750 nm) through controlled printing loops.
- Ligand Solution Preparation: Prepare a 0.2% (v/v) solution of ethanedithiol (EDT) in acetonitrile as the exchange medium.
- Exchange Process: Apply EDT solution to the printed QD microstructures for 60 seconds. This duration achieves approximately 90% replacement of native oleic acid ligands without structural damage.
- Rinsing and Drying: Rinse thoroughly with pure acetonitrile to remove displaced ligands and residual exchange solution. Dry under nitrogen flow. Critical Note: Prolonged exposures (>1 hour) cause systematic degradation of microstructures through QD removal, regulated by surface-to-bulk ratios and solvent interactions [25].
Sequential Ligand Exchange for Polymer:QD Bulk Heterojunctions
This method preserves film morphology while enhancing electronic coupling in polymer-QD blends for photovoltaic applications [26]:
- Initial Solution-Phase Exchange: Synthesize OA-capped PbS QDs following the Hines and Scholes method. Perform first ligand exchange by treating QD solution with butylamine (BA) to replace OA partially.
- Film Deposition: Blend BA-capped PbS QDs with PTB1 polymer in 1:9 weight ratio and deposit via solution casting to form a bulk heterojunction film.
- Solid-State Secondary Exchange: Treat the deposited film with 3-mercaptopropionic acid (MPA) solution to replace remaining long ligands with short thiols.
- Morphological Validation: Use scanning transmission electron microscopy (STEM) and tomographic reconstruction to verify preservation of nanoscale phase separation without void formation. Advantage: This sequential approach prevents the severe morphological changes and void defects observed when directly exchanging OA with MPA in solid films [26].
Sulfide Capping for InSb Quantum Dot Photodiodes
This protocol enhances carrier mobility in III-V QD-based short-wavelength infrared photodiodes [23]:
- QD Synthesis: Synthesize InSb CQDs using oleylamine as solvent and super hydride as reducing agent, with optimized In/Sb precursor ratio and SH concentration to suppress oxidation.
- Size Selection: Perform four-step centrifugation with incremental methanol addition to narrow size distribution, collecting fractions A-D for optimal monodispersity.
- Sulfide Ligand Exchange: Replace native oleylamine ligands with sulfide ions using appropriate sulfur precursors.
- Film Characterization: Confirm reduced interparticle distance (1.5 ± 0.5 nm vs. 5.0 ± 0.5 nm for OLA-capped) through HR-TEM and GI-XRD.
- Device Fabrication: Assemble photodiode architecture with sulfide-capped InSb CQDs as photoactive layer. Measure electrical properties showing low dark current density (~nA cm⁻²) and improved EQE (18.5%) at 0V bias.
Visualization of Ligand Exchange Processes
Diagram 1: Ligand exchange process showing the transition from long insulating ligands to short conducting ligands, resulting in reduced interparticle distance and enhanced carrier mobility.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Conventional Ligand Exchange Processes
| Reagent Category | Specific Examples | Function & Mechanism | Compatible QD Systems |
|---|---|---|---|
| Inorganic Ligands | Sulfide salts, Indium chloride (InCl₃), Halide salts (Br⁻, I⁻) | Atomic-scale bridging; Anionic site passivation; Z-type ligand exchange | InSb, InP, PbS, CdSe [23] [27] |
| Short-Chain Thiols | Ethanedithiol (EDT), 3-Mercaptopropionic acid (MPA) | Strong covalent binding to metal sites; Radical-mediated exchange processes | PbS, CdSe, ZnO [25] [26] |
| Short-Chain Amines | Butylamine (BA), Oleylamine (OLA) | L-type ligand binding; Intermediate for sequential exchanges; Size-selective precipitation | PbS, InSb, Perovskite QDs [23] [26] |
| Native Ligands | Oleic acid (OA), Tri-n-octylphosphine oxide (TOPO) | Initial stabilization; High-temperature synthesis compatibility | Most II-VI and III-V QD systems [23] [24] |
| Solvents | Tetradecane, Acetonitrile, Toluene, Dichloromethane | Ink formulation (non-polar); Exchange medium (polar); Purification and washing | System-dependent [25] [26] |
Conventional ligand exchange strategies centered on shortening the organic bridge between quantum dots have proven highly effective for enhancing charge transport in QD solids. The methodologies detailed herein—from solid-state exchanges to sequential processing approaches—provide researchers with robust tools for engineering QD films with improved electronic properties. As the field advances, emerging ligand design concepts including redox-active molecules and conjugated organic systems suggest a shift from viewing ligands as passive spacers to treating them as active components in charge transport pathways. Nevertheless, the fundamental principle of reducing interparticle tunneling barriers through strategic ligand selection remains paramount for developing next-generation QD optoelectronic devices.
The pursuit of efficient charge transport in quantum dot (QD) solids represents a central challenge in advancing next-generation optoelectronic devices. For over two decades, scientific understanding and engineering approaches have primarily focused on using ligands as passive spacers, where their primary function was to modulate the interparticle distance and thus lower the tunneling barrier for electron hopping between adjacent QDs [4]. In this conventional paradigm, shorter ligands generally enhance conductivity by reducing the distance carriers must tunnel, while longer, insulating ligands hinder transport. However, a transformative paradigm shift is emerging, moving beyond this passive role toward a design strategy where ligands function as active electronic components in the transport process. This paradigm, known as the redox ligand paradigm, utilizes molecular functionalities that introduce defined electronic states into the QD solid, thereby opening additional pathways for charge propagation [4]. Among the most promising candidates for this role are ferrocene derivatives, whose well-defined redox chemistry and molecular structure enable them to act as active mediators for long-range charge transport, fundamentally changing the conductive properties of quantum dot assemblies.
The Ferrocene Advantage: An Ideal Redox-Active Scaffold
Ferrocene (Fc), an organometallic complex with an iron center situated between two cyclopentadienyl rings, possesses a unique combination of properties that make it exceptionally suitable for application as a redox-active ligand in QD solids. Its well-behaved, reversible, one-electron outer-sphere redox couple (Fc/Fc+) provides a stable and predictable energy level for mediating charge transfer [4]. Furthermore, its functionalizability allows for the incorporation of anchoring groups, such as carboxylates, which enable covalent attachment to the QD surface. The recent groundbreaking synthesis of a stable 20-electron ferrocene derivative further expands the potential chemical and redox behaviors of these complexes, challenging the long-standing 18-electron rule and unlocking new possibilities in catalysis and materials science [28]. This expansion of accessible oxidation states suggests that ferrocene derivatives could facilitate a wider range of electron transfer processes than previously thought possible, enhancing their utility as charge transport mediators.
Charge Transport Mechanisms in QD-Redox Ligand Assemblies
In a QD solid functionalized with redox ligands, charge transport is no longer limited to a single pathway. Research on ZnO QDs with ferrocene carboxylate (FcCOO–) ligands has demonstrated that two distinct, complementary mechanisms operate concurrently.
- Electron Hopping through the Quantum Dot Conduction Band: This is the traditional pathway, where electrons tunnel from the conduction band of one QD to the conduction band of a neighboring QD. The rate of this process is exponentially dependent on the inter-dot distance and is thus influenced by the physical size of the ligands [4].
- Self-Exchange via the Redox Ligands: This is the novel pathway enabled by the redox-active ligands. In this mechanism, an electron can "hop" between adjacent ferrocene centers. A reduced ferrocene (Fc) can transfer an electron to an adjacent oxidized ferrocenium (Fc+), effectively propagating the charge through the ligand network without direct involvement of the QD cores. This self-exchange reaction creates a chain reaction that can facilitate long-range charge transport across the entire film [4].
The overall charge transport through the assembly is thus a complex interplay of these two pathways, accompanied by necessary charge transfer between the QDs and redox ligands, as well as ion transport to maintain charge neutrality within the porous film [4].
Quantitative Data: The Impact of Ligand Engineering
The performance of QD solids is profoundly affected by both the chemical nature and the physical dimensions of the surface ligands. The following tables summarize key quantitative findings from research on ligand engineering.
Table 1: Impact of Different Ligand Treatments on Inter-Dot Distance in PbS QD Films [29]
| Ligand Treatment | Chemical Description | Average Inter-Dot Distance (nm) | Distance Reduction vs. OA (nm) |
|---|---|---|---|
| Oleic Acid (OA) | Long aliphatic ligand (C18) | ~10.2 | (Baseline) |
| 1,3-BDT | Conjugated dithiol, meta configuration | 9.3 ± 0.8 | 0.9 |
| EDT | Short-chain dithiol (C2 backbone) | 7.8 ± 0.8 | 2.4 |
| MPA | Short-chain with thiol & carboxylate (C2) | 7.6 ± 0.8 | 2.6 |
| (NH₄)₂S | Inorganic sulfide linker | 6.7 ± 0.7 | 3.5 |
Table 2: Charge Transport Properties of ZnO-FcCOO– QD Assemblies [4]
| Property | Description | Value / Observation |
|---|---|---|
| QD Core Material | - | ZnO |
| Redox Ligand | - | Ferrocene Carboxylate (FcCOO⁻) |
| Ligand Coverage | Estimated from XPS data | ~10 ligands per QD |
| Primary Transport Pathways | Identified via electrochemistry | 1. Electron hopping via QD CB2. Self-exchange via redox ligands |
| Kinetic Dependence | How transport rates vary with ligand coverage | Electron hopping & ion transport: Independent of FcCOO⁻ concentrationSelf-exchange: Strongly dependent on FcCOO⁻ concentration (percolation theory) |
The data in Table 1 clearly demonstrates how ligand choice directly controls inter-dot spacing. Shorter ligands like EDT and MPA bring QDs significantly closer, facilitating better tunneling. The (NH₄)₂S treatment, which creates an inorganic sulfide bridge, results in the smallest separation and even partial fusion of QDs [29]. This control over inter-dot distance remains a critical factor for the electron hopping component of transport, even in active ligand systems.
Experimental Protocols: Methodology for Investigating Redox-Ligand Systems
To ensure reproducibility and provide a clear technical roadmap, this section outlines key experimental procedures for creating and characterizing QD/redox-ligand assemblies.
Synthesis and Functionalization of ZnO-Ferrocene Carboxylate QDs
- QD Synthesis: Synthesize ZnO QDs according to established methods to achieve a bandgap of approximately 3.86 eV, corresponding to a diameter of ~2.5 nm [4]. This can be confirmed via steady-state UV-Vis spectroscopy.
- Film Preparation: Drop-cast the synthesized ZnO QD dispersion onto a clean, conductive ITO-coated glass substrate to form a porous film with a thickness of approximately 1.5 μm, as verified by profilometry [4].
- Ligand Exchange via Post-Assembly Functionalization: Immerse the ZnO QD film into a solution of ferrocene carboxylic acid (FcCOOH) in acetonitrile. Allow the immersion to proceed overnight to ensure complete ligand exchange. Following functionalization, rinse the film thoroughly with pure acetonitrile to remove any unbound, free FcCOOH molecules [4].
- Validation of Binding: Confirm successful anchoring and distribution of FcCOO– ligands using a combination of techniques:
- X-ray Photoelectron Spectroscopy (XPS) Depth-Profiling: To verify a uniform distribution of iron (Fe) and zinc (Zn) throughout the film depth, with a consistent Zn/Fe atomic ratio indicating ~10 ligands per QD [4].
- Nuclear Magnetic Resonance (NMR) Spectroscopy: To demonstrate the binding of ligands, evidenced by broadened NMR peaks for the ferrocene protons due to restricted molecular tumbling, and the absence of sharp peaks from unbound FcCOOH [4].
Electrochemical Interrogation of Charge Transport Pathways
The electronic properties and charge transfer kinetics of the QD/redox-ligand assembly can be probed electrochemically.
- Setup: Use a standard three-electrode electrochemical cell with the QD film on ITO as the working electrode, a platinum counter electrode, and a suitable reference electrode (e.g., Ag/AgCl). The electrolyte is typically a solution of tetrabutylammonium hexafluorophosphate (TBAPF₆) in acetonitrile [4].
- Cyclic Voltammetry (CV): Perform CV scans, for example, from -0.6 V to -1.7 V and then to +0.9 V (vs. Fc/Fc+), to obtain the "fingerprint" of the assembly. This technique allows for the identification of distinct signals corresponding to different processes [4]:
- Signal (i): A broad signal during the initial negative scan, corresponding to charge injection into the conduction band of the ZnO QDs.
- Signal (ii): A quasi-reversible peak at more positive potentials, corresponding to the redox activity of the ferrocene/ferrocenium couple, indicative of the self-exchange pathway.
Visualizing the Paradigm: Pathways and Workflows
The following diagrams, generated using Graphviz, illustrate the core concepts and experimental workflows of the redox ligand paradigm.
Charge Transport Pathways in a QD-Redox Ligand Solid
Experimental Workflow for Fabrication and Analysis
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for QD-Redox Ligand Research
| Reagent / Material | Function in Research | Technical Notes |
|---|---|---|
| Zinc Oxide QDs | Model quantum dot core for building the assembly. | ~2.5 nm diameter, bandgap ~3.86 eV. Provide a stable, electrochemically addressable platform [4]. |
| Ferrocene Carboxylic Acid (FcCOOH) | Precursor for the redox-active ligand. | The carboxylate group anchors to metal oxide surfaces; the ferrocene moiety provides the redox-active state for self-exchange [4]. |
| Tetrabutylammonium Hexafluorophosphate (TBAPF₆) | Electrolyte salt. | Provides mobile ions (TBA⁺, PF₆⁻) in the electrolyte solution necessary for charge compensation during electrochemical processes [4]. |
| Acetonitrile (MeCN) | Solvent for ligand exchange and electrochemistry. | A polar aprotic solvent with a wide electrochemical window, ideal for non-aqueous electrochemistry [4]. |
| Indium-Tin-Oxide (ITO) Glass | Conductive substrate. | Serves as the working electrode for electrochemical measurements and a physical support for the QD film [4]. |
| 1,2-Ethanedithiol (EDT) | Short, passive ligand for comparative studies. | A common short ligand used to reduce inter-dot distance and study passive tunneling transport [29]. |
The redox ligand paradigm, exemplified by the use of ferrocene derivatives, marks a significant departure from traditional ligand engineering in quantum dot solids. By introducing active electronic states rather than acting as mere passive spacers, this approach provides a powerful and versatile tool for controlling charge transport properties. The ability to orchestrate multiple charge transport pathways—specifically QD band hopping and ligand-mediated self-exchange—offers unprecedented tunability for the design of advanced optoelectronic materials.
Future progress in this field will likely be driven by the synthesis of novel redox ligands with tailored energy levels and multiple redox states, inspired by breakthroughs such as the 20-electron ferrocene derivatives [28]. Furthermore, combining this paradigm with other advanced strategies, like inorganic ligand exchange with (NH₄)₂S to minimize inter-dot spacing, could lead to synergistic effects and record-performing materials [29]. As the fundamental understanding of charge transport mechanisms in these complex assemblies deepens, the rational design of QD solids with bespoke electronic properties for applications in photovoltaics, catalysis, and sensing will become increasingly feasible, solidifying the redox ligand paradigm as a cornerstone of next-generation materials science.
Colloidal quantum dot (CQD) films represent a promising class of materials for next-generation optoelectronic devices, including photovoltaics, photodetectors, and light-emitting diodes. Their appeal lies in large-area solution processability and bandgap tunability through the quantum size effect [30]. However, the high surface-to-volume ratio of CQDs makes these films highly susceptible to trap state densities when surfaces are imperfectly passivated. These trap states promote charge carrier recombination, which is detrimental to device performance [30] [31]. The central challenge lies in balancing two competing needs: long insulating ligands that provide excellent colloidal stability during synthesis and processing, versus short conductive ligands that enable efficient charge transport in solid films.
Surface ligand engineering has emerged as the pivotal strategy for mitigating loss processes in quantum dot solar cells [3]. Ligands maintain the shape and size of individual dots in solid films, preserve the clean energy bandgap of individual particles, and critically control charge carrier conduction across the solid film [3]. This technical guide examines hybrid passivation strategies that combine solution-phase and solid-state exchange approaches to achieve optimal surface passivation and charge transport properties in quantum dot solids, with particular focus on implications for PbS CQD-based optoelectronic devices.
The Ligand Length Dilemma: Charge Transport versus Passivation Quality
The performance of quantum dot solids is fundamentally governed by surface chemistry. Initial synthetic routes produce CQDs capped with long-chain insulating ligands (e.g., oleic acid), which provide excellent colloidal stability and monodispersity but create significant barriers to charge transport in solid-state films [3] [31]. The subsequent ligand exchange process, where these long-chain ligands are replaced with shorter alternatives, thus becomes a critical step in device fabrication.
Impact of Ligand Characteristics on Electronic Properties
Research demonstrates that changes in the size, shape, and functional groups of small-chain organic ligands directly modulate mobility, dielectric constant, and carrier doping density in PbS quantum dot solids [3]. These parameters in turn govern performance, stability, and recombination processes in photovoltaic devices. Short ligands reduce inter-dot spacing, facilitating enhanced wavefunction overlap between neighboring dots and significantly improving charge carrier mobility. However, incomplete surface coverage or poorly coordinated short ligands can create electronic trap states that promote non-radiative recombination, offsetting the benefits of reduced inter-dot spacing [3] [31].
Limitations of Single-Method Passivation Approaches
Conventional solid-state ligand exchange processes, while widely implemented, suffer from significant limitations. Studies using tetrabutylammonium iodide (TBAI) treatments revealed that the initial long-chain capping ligands cannot be fully replaced within practical time scales, resulting in low carrier mobility and disordered packing densities [31]. Furthermore, halide-only passivation often leaves charged Pb atoms that generate sub-bandgap states, contributing to voltage deficits in solar cells [31]. Solution-based ligand exchange approaches offer superior control over surface charge balance but present challenges in maintaining film morphology and achieving complete ligand coverage when used alone [30].
Hybrid Passivation: Principles and Methodologies
Hybrid passivation addresses the limitations of single-method approaches by combining the complementary strengths of solution-phase and solid-state exchange techniques. This integrated strategy enables more comprehensive surface coverage and superior trap state passivation.
Fundamental Mechanism
The hybrid passivation approach employs a dual-ligand system typically consisting of halide anions combined with short organic molecules. This creates a robust passivation scheme where each ligand type addresses different surface trap sites [30] [31]. Halide ions (I⁻, Br⁻, Cl⁻) provide strong coordination with Pb atoms on the CQD surface according to the Hard and Soft Acids and Bases (HSAB) theory, where Pb²⁺ acts as a soft acid that preferentially binds with soft bases like I⁻ [31]. Concurrently, small organic molecules such as pyridine access and passivate trap sites that may be sterically inaccessible to larger halide ions or where the binding chemistry is more favorable [30].
Table 1: Key Components in Hybrid Passivation Systems
| Component Type | Specific Examples | Primary Function | Impact on Device Performance |
|---|---|---|---|
| Halide Anions | I⁻ (from TBAI), Br⁻, Cl⁻ | Primary surface passivation via Pb coordination | Reduces trap states, improves VOC |
| Organic Ligands | Pyridine, 3-Mercaptopropionic acid | Access sterically hindered sites, secondary passivation | Improves JSC, enhances film morphology |
| Lead Precursors | PbO, Pb acetate | Quantum dot core formation | Determines initial dot quality and size distribution |
| Sulfur Precursors | Bis(trimethylsilyl)sulfide (TMS) | Quantum dot core formation | Controls reaction kinetics and size distribution |
Experimental Protocol: Implementing Hybrid Passivation
The following detailed methodology outlines a representative hybrid passivation process for PbS CQDs, synthesized from established protocols in the literature [30] [31].
Initial PbS CQD Synthesis (Heat Injection Method)
Reagent Preparation:
- Create lead precursor by combining 0.45 g PbO, 1.5 mL oleic acid, and 20 mL 1-octadecene in a three-neck flask.
- Prepare sulfur precursor by dissolving 0.1 mL bis(trimethylsilyl)sulfide (TMS) in 10 mL 1-octadecene.
Reaction Process:
- De-gas the lead precursor mixture under vacuum at 90°C for 60 minutes.
- Heat the solution to 120°C under N₂ atmosphere until the PbO completely dissolves, forming a clear solution.
- Rapidly inject the sulfur precursor solution into the reaction vessel.
- Immediately quench the reaction after 60 seconds using an ice-water bath.
Purification:
- Precipitate the nanocrystals using anhydrous acetone and centrifuge at 7500 rpm for 5 minutes.
- Re-disperse the pellet in toluene and precipitate again with acetone.
- Repeat this washing process three times total.
- Finally, disperse the purified PbS CQDs in octane at a concentration of 50 mg/mL for storage.
Hybrid Passivation Procedure
Solution-Phase Pre-Treatment:
- Dissolve the pristine PbS CQDs in anhydrous toluene to create a 25 mg/mL solution.
- Add a 1:5 molar ratio (relative to Pb sites) of pyridine to the CQD solution and stir for 12 hours at 50°C.
- Precipitate the partially exchanged CQDs with acetone and centrifuge.
- Re-disperse the CQDs in butanol for film deposition.
Film Formation and Solid-State Exchange:
- Spin-coat the pre-treated CQD solution onto the substrate at 2000 rpm for 30 seconds.
- Immerse the film immediately in a 10 mg/mL solution of TBAI in methanol for 30 seconds.
- Rinse thoroughly with pure methanol to remove excess ligands and byproducts.
- Repeat the layering process (spin-coating followed by solid-state exchange) 8-10 times to achieve the desired film thickness (~300 nm).
Post-Treatment:
- Anneal the completed film at 70°C for 30 minutes under N₂ atmosphere to improve inter-dot coupling and remove residual solvent.
Figure 1: Hybrid Passivation Experimental Workflow
Comparative Performance Analysis of Passivation Strategies
The effectiveness of hybrid passivation becomes evident when comparing the structural, electronic, and device-level performance metrics against single-method approaches.
Structural and Morphological Characterization
Atomic force microscopy (AFM) and transmission electron microscopy (TEM) analyses reveal striking differences between TBAI-only and hybrid-passivated PbS CQD films. TBAI-only treated films exhibit non-uniform surfaces with significant cracks and high roughness, indicating insufficient ligand removal and quantum dot aggregation [31]. In contrast, hybrid-passivated films show remarkably uniform surfaces with minimal cracking and reduced roughness, suggesting improved quantum dot packing and maintained quantum confinement [31]. High-resolution TEM further confirms that hybrid treatment preserves individual PbS CQDs with distinguishable crystal boundaries, while TBAI-only treatment leads to large fused crystal domains (10-50 nm) that diminish quantum confinement effects [31].
Fourier-transform infrared (FT-IR) spectroscopy provides chemical evidence for the superiority of hybrid passivation. The characteristic symmetric (2854 cm⁻¹) and asymmetric (2924 cm⁻¹) stretching vibrations of -CH₂ from oleic acid ligands nearly completely disappear after hybrid passivation, while these peaks persist in TBAI-only treated films, confirming more complete removal of initial ligands in the hybrid approach [31].
Electronic Properties and Device Performance
The hybrid passivation strategy demonstrates significant advantages in electronic properties and ultimate device performance, particularly in solar cell applications.
Table 2: Performance Comparison of Passivation Strategies in PbS CQD Solar Cells
| Parameter | TBAI-only Passivation | Hybrid (TBAI+Pyridine) Passivation | Measurement Significance |
|---|---|---|---|
| Power Conversion Efficiency (PCE) | 5.3% [31] | 6.8% [31] | Overall device performance metric |
| Certified Efficiency Record | - | 7.0% [30] | Independent verification of performance |
| Film Roughness (RMS) | Higher, non-uniform [31] | Lower, flat and uniform [31] | Morphological quality indicator |
| Ligand Removal Efficiency | Incomplete (FT-IR detectable OA) [31] | Near-complete OA removal [31] | Surface passivation completeness |
| Trap State Density | Higher, significant sub-gap states [31] | Substantially reduced [30] [31] | Electronic quality metric |
| Charge Extraction Efficiency | Limited by trap states [31] | Enhanced [31] | Carrier collection capability |
The enhancement in power conversion efficiency from 5.3% to 6.8% with hybrid passivation stems from improvements across all major solar cell parameters [31]. The open-circuit voltage (VOC) increases due to reduced trap-assisted recombination, while the short-circuit current density (JSC) improves through enhanced charge extraction efficiency. The fill factor also benefits from reduced series resistance and improved film morphology [31].
Advanced Hybrid Passivation Systems and Future Directions
Beyond the basic halide-organic ligand combinations, research continues to develop increasingly sophisticated hybrid passivation approaches to further optimize CQD device performance.
Emerging Hybrid Strategies
Recent advances include atomic ligand passivation combined with organic crosslinkers, which provides more comprehensive surface coverage [32]. Additionally, researchers are exploring sequential solution-phase exchanges with different ligand types before solid-state treatment to address specific surface sites more strategically. Another promising direction involves the incorporation of bifunctional ligands that simultaneously passivate surface defects and promote inter-dot coupling through π-conjugated systems [3].
Research Reagent Solutions for Hybrid Passivation
Table 3: Essential Research Reagents for Hybrid Passivation Studies
| Reagent Category | Specific Examples | Function in Hybrid Passivation | Technical Considerations |
|---|---|---|---|
| Halide Salts | Tetrabutylammonium iodide (TBAI), PbI₂, NH₄I | Provide halide anions for primary surface passivation | Solubility in polar solvents (methanol, acetone) critical for solid-state exchange |
| Short-Chain Organic Ligands | Pyridine, 3-Mercaptopropionic acid (3-MPA), Ethanedithiol (EDT) | Secondary passivation and steric access | Molecular length and binding group affinity determine effectiveness |
| Solvents | Octane, Toluene, Methanol, Butanol, Acetone | Dispersion, processing, and purification medium | Purity and anhydrous conditions essential for reproducible results |
| Lead Precursors | Lead oxide (PbO), Lead acetate | Determine initial quantum dot size and quality | Impact reaction kinetics and final size distribution |
| Sulfur Precursors | Bis(trimethylsilyl)sulfide (TMS), Elemental sulfur | Control quantum dot nucleation and growth | TMS enables high-temperature synthesis for narrow size distributions |
Figure 2: Surface Passivation Transition in Hybrid Strategy
Hybrid passivation strategies that strategically combine solution-phase and solid-state ligand exchange represent a significant advancement in quantum dot surface engineering. By leveraging the complementary strengths of both approaches—solution-phase control over surface charge balance and solid-state efficiency in film formation—this methodology enables more complete surface coverage and superior trap state passivation compared to either method alone. The resulting enhancement in charge transport properties, coupled with reduced non-radiative recombination, directly translates to improved performance in optoelectronic devices, as evidenced by certified solar cell efficiencies reaching 7.0% [30].
The fundamental principles underlying hybrid passivation align directly with the broader thesis that surface ligand length profoundly affects charge transport in quantum dot solids. While shorter ligands generally enhance inter-dot coupling and charge mobility, they risk incomplete surface coverage if deployed improperly. The hybrid approach resolves this dichotomy by employing a coordinated ligand system where different ligand types address specific surface sites and functional requirements. As research progresses, further refinement of these strategies—including development of novel ligand combinations and optimized processing sequences—will continue to push the performance boundaries of quantum dot-based optoelectronic devices, potentially enabling their commercialization in various technological applications.
Surface ligand engineering serves as a critical tool for modulating the charge transport properties of quantum dot (QD) solids, a fundamental aspect for advancing optoelectronic devices. This whitepaper explores this principle through two specific case studies: the use of lead iodide (PbI₂) as a precursor and passivant in perovskite structures, and the application of benzylammonium ligands in perovskite nanocrystal (LHP NC) light-emitting diodes (LEDs). The document synthesizes current research to demonstrate how atomic and molecular ligands directly influence electronic coupling, trap state density, and overall device performance. Structured quantitative data, detailed experimental protocols, and mechanistic diagrams are provided to serve as a reference for researchers developing next-generation QD-based technologies.
In quantum dot solids, the organic ligand shell that stabilizes individual nanocrystals in solution becomes a primary determinant of interparticle spacing and electronic coupling in the solid state. Long, insulating aliphatic ligands (e.g., oleic acid) impose large potential barriers between QDs, forcing charge carriers to traverse films via slow, thermally activated hopping [32] [1]. Shortening or replacing these ligands is therefore a central strategy for enhancing charge carrier mobility.
The relationship between ligand structure and function can be understood through two primary mechanisms:
- Interparticle Tunneling: Shorter ligands reduce the physical distance between QD cores, exponentially increasing the probability of electron tunneling between adjacent dots [1] [5].
- Active Transport Pathways: Certain ligands, such as those with conjugated systems or redox-active moieties, can introduce electronic states that actively mediate charge transport rather than acting as passive spacers [33] [4].
This guide examines these concepts through the lens of two specific materials: the atomic ligand in PbI₂-derived perovskites and the molecular benzylammonium ligand.
Case Study 1: Lead Iodide (PbI₂) - Precursor and Passivant
Lead iodide is a foundational material in metal halide perovskite photovoltaics, serving both as a direct precursor and as a source of passivating halide ions.
Material Properties and Function
Table 1: Key Properties of Lead Iodide (PbI₂) [34] [35]
| Property | Value | Relevance to Device Performance |
|---|---|---|
| Chemical Formula | PbI₂ | Precursor for perovskite synthesis (e.g., CH₃NH₃PbI₃) |
| Crystal Structure | Rhombohedral, hexagonal | Template for perovskite crystal growth |
| Band Gap | 2.34 eV (direct) [34] | Determines optical absorption and electronic characteristics |
| Solubility | Low in cold water; increases with temperature | Enables specific crystallization methods (e.g., "golden rain") |
| Hole Mobility | ≤ 2 cm² V⁻¹ s⁻¹ [35] | Intrinsic charge transport property |
| Electron Mobility | ≤ 8 cm² V⁻¹ s⁻¹ [35] | Intrinsic charge transport property |
PbI₂'s role extends beyond that of a simple precursor. In colloidal quantum dot (CQD) systems, iodide ions (I⁻) from sources like tetrabutylammonium iodide (TBAI) are used in ligand exchange to passivate the surfaces of PbS CQDs. The I⁻ ion binds strongly to undercoordinated Pb atoms on the CQD surface, effectively reducing the density of charge trap states [5] [36]. This atomic ligand is significantly shorter than native organic ligands, leading to enhanced interdot electronic coupling.
Quantitative Performance Impact of Halide Ligand Exchange
Ligand exchange with halide ions is a well-established method for improving the performance of QD solar cells. The data below compares the performance of PbS CQD solar cells treated with different passivation strategies.
Table 2: Performance of PbS CQD Solar Cells with Different Passivation Methods [5]
| Passivation Strategy | Power Conversion Efficiency (PCE) | Key Film Characteristics | Implication for Charge Transport |
|---|---|---|---|
| TBAI-only | 5.3% | Non-uniform films, significant cracking, aggregated CQDs | Limited charge extraction due to film disorder and residual traps |
| Hybrid (TBAI + Pyridine) | 6.8% | Uniform, flat films with high packing density and fewer cracks | Reduced trap states and shorter ligand length enhance mobility and collection |
The superior performance of the hybrid passivation is attributed to the synergistic effect of the atomic iodide ligand and the short, complexing pyridine molecule. Pyridine aids in the near-complete removal of pristine bulky ligands and provides additional surface coordination, further reducing surface defects [5].
Experimental Protocol: Solid-State Ligand Exchange with Halide Ions
This protocol is widely used for fabricating high-performance PbS CQD solar cell active layers [36].
- Film Fabrication: Spin-coat a solution of PbS CQDs capped with long-chain oleic acid ligands in a non-polar solvent (e.g., octane) onto a substrate to form an as-cast film.
- Ligand Introduction: Introduce a solution of the new ligand (e.g., TBAI in methanol, typically 0.01-0.02 M) onto the CQD film.
- Soaking and Exchange: Allow the film to soak for 20-60 seconds to enable the exchange reaction, where I⁻ displaces the oleate ligands.
- Solution Removal: Spin-coat the film to remove the excess ligand solution and displaced oleic acid.
- Washing: Rinse the film with a neat polar solvent (e.g., methanol) to remove any remaining reaction by-products and excess ligands.
- Layer Buildup: Repeat steps 1-5 in a layer-by-layer fashion to build up the desired film thickness (e.g., 300-400 nm for a solar cell).
Case Study 2: Benzylammonium - A Molecular Ligand for Enhanced Performance
Benzylammonium (BA) is an example of a molecular ligand where its aromatic structure actively contributes to charge transport.
Mechanism of Action
In perovskite nanocrystal (LHP NC) LEDs, benzylammonium ligands enhance performance through several mechanisms [33]:
- Orbital Overlap: The π-electrons of the ligand's aromatic ring interact with the orbitals on the NC surface. This conjugated structure enhances wavefunction delocalization, facilitating charge injection from the electrodes into the NC and improving transport between NCs.
- Trap State Passivation: The ammonium group (-NH₃⁺) effectively binds to surface sites, reducing non-radiative recombination centers and improving photoluminescence quantum yield (PLQY).
- Stability: The ligand contributes to the overall stability of the NCs, which is crucial for device longevity.
Quantitative Performance Impact in Perovskite LEDs
The effect of benzylammonium halide ligand exchange on the performance of CsPbBr₃ NC LEDs is significant, as shown in the comparative data below.
Table 3: Performance of CsPbBr₃ NC LEDs with and without Benzylammonium Ligand Exchange [33]
| Device Type | External Quantum Efficiency (EQE) | Maximum Current Efficiency (CEmax) | Key Improvement Factors |
|---|---|---|---|
| Pristine NCs | 2.4% | 7.8 cd A⁻¹ | Baseline |
| BA Bromide-treated | 5.88% | 19.5 cd A⁻¹ | Enhanced charge injection, reduced defects, improved PLQY |
| BA Chloride-treated | 5.50% | 16.6 cd A⁻¹ | Enhanced charge injection, reduced defects, improved PLQY |
Experimental Protocol: Ligand Exchange with Benzylammonium on Perovskite NCs
This procedure outlines the post-synthetic treatment of CsPbBr₃ NCs to incorporate benzylammonium ligands [33].
- NC Synthesis: Synthesize high-quality CsPbBr₃ NCs using standard hot-injection methods, initially capped with traditional aliphatic ligands (e.g., oleylammonium).
- Ligand Solution Preparation: Prepare a solution of the benzylammonium halide salt (e.g., benzylammonium bromide or chloride) in a solvent such as isopropanol or butanol.
- Purification: Purify the pristine NCs to remove excess initial ligands and reaction residues.
- Exchange Reaction: Re-disperse the purified NCs in an apolar solvent (e.g., hexane or toluene) and add the benzylammonium halide solution under stirring. The reaction may be facilitated by mild heating.
- Purification: Precipitate the ligand-exchanged NCs by adding a polar anti-solvent (e.g., ethyl acetate), then isolate them via centrifugation.
- Washing and Dispersion: Re-disperse the final NC pellet in an appropriate solvent for device fabrication (e.g., octane for spin-coating).
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Ligand Engineering in Quantum Dot Research
| Reagent | Function | Example Use Case |
|---|---|---|
| Lead Iodide (PbI₂) | Precursor material; source of passivating iodide ions | Formation of methylammonium lead iodide perovskite; halide ligand exchange on PbS CQDs [34] [5] |
| Tetrabutylammonium Iodide (TBAI) | Provides mobile I⁻ ions for atomic ligand exchange | Solid-state ligand exchange to passivate PbS CQD surfaces [5] [36] |
| Benzylammonium Halides | Molecular ligands with conjugated aromatic rings | Ligand exchange on perovskite NCs to improve charge injection and transport in LEDs [33] |
| Pyridine | Short, bidentate ligand that strongly coordinates to Pb atoms | Hybrid passivation with TBAI to remove bulky ligands and reduce surface defects on PbS CQDs [5] |
| 1,2-Ethanedithiol (EDT) | Short-chain dithiol ligand for cross-linking QDs | Creating stable, conductive p-type layers in CQD solar cells [36] |
| Dimethyl Sulfoxide (DMSO) | Solvent with high coordinating ability to Pb²⁺ | Influencing crystallization and structural topology in perovskite precursor solutions [37] |
Diagrams of Charge Transport Mechanisms
The following diagrams illustrate the fundamental concepts of charge transport in ligand-capped quantum dot solids.
Charge Transport in QD Solids
Active Transport via Redox Ligands
The case studies of lead iodide and benzylammonium ligands underscore a fundamental principle in quantum dot solid research: precise control over surface chemistry is as critical as control over the nanocrystal core itself. The transition from viewing ligands as passive stabilizers to recognizing them as active components in charge transport has opened new avenues for device optimization.
- For Atomic Ligands (PbI₂/I⁻): The primary benefit lies in dramatic reductions in interparticle spacing and effective passivation of anionic surface traps, leading to significant gains in conductivity and photovoltaic performance [5] [36].
- For Molecular Ligands (Benzylammonium): The advantage extends beyond spatial considerations to include the introduction of novel electronic pathways via conjugation, which enhances charge injection and transport in light-emitting devices [33] [4].
Future research directions will likely focus on the design of increasingly sophisticated multifunctional ligands that combine short length, robust passivation, and active transport properties, while also addressing challenges in scalability and long-term operational stability. The integration of computational screening with synthetic efforts will be key to accelerating the discovery of next-generation ligand systems.
In quantum dot (QD) solids, surface ligands are more than mere stabilizers; they are a critical design element that directly governs charge transport, a fundamental process in electronic and optoelectronic devices. The core thesis driving contemporary research is that strategic ligand engineering—controlling their length, chemistry, and functionality—can systematically tune the electronic coupling between QDs, thereby dictating the performance of devices like transistors and solar cells. By moving beyond the traditional view of ligands as passive spacers to actively engineering them as conductive elements or dipole-modulating layers, researchers are unlocking new frontiers in QD-based device efficiency and functionality [1] [4]. This guide delves into the mechanisms, experimental data, and protocols that underpin this transformative approach.
Fundamental Mechanisms of Ligand-Mediated Charge Transport
The electronic properties of a QD solid are not merely an average of its individual components. They emerge from the complex interplay of quantum confinement and interparticle coupling, which is predominantly modulated by the ligand shell [1].
The Tunneling Barrier: Ligand Length and Electronic Coupling
The primary role of a ligand in charge transport is to define the potential energy barrier between adjacent QDs. Charge carriers typically traverse the solid via tunneling or hopping mechanisms, and the rate of this process depends exponentially on the inter-dot distance. Short ligands reduce this distance, dramatically increasing the tunneling probability and thus the charge carrier mobility [1] [36]. For instance, replacing long oleic acid ligands (∼1.8 nm) with shorter n-butylamine ligands (∼0.6 nm) can increase the electrical conductivity of a PbS QD solid by a factor of 180 [16].
Beyond Spacers: Active Transport and Energy Level Tuning
Modern ligand engineering introduces more sophisticated functions:
- Active Redox Ligands: Ligands with electronic states, such as ferrocene carboxylate, can create an alternative charge transport pathway via a self-exchange chain reaction. This provides a complementary route to conduction via the QDs' conduction band, actively engineering long-range transport [4].
- Band Alignment Control: Ligands induce surface dipoles that shift the QDs' energy levels. For example, PbS QDs treated with tetrabutylammonium iodide (TBAI) exhibit a deeper work function than those treated with 1,2-ethanedithiol (EDT). This allows for the design of hole-extraction and electron-blocking layers within a QD solar cell, improving charge collection [38].
- Trap State Passivation: Certain ligands, particularly halides or tailored organic molecules, effectively passivate surface defects (trap states). This reduces charge carrier recombination, a critical factor for enhancing the open-circuit voltage in solar cells and the on/off ratio in transistors [39] [1].
The following diagram illustrates the primary charge transport pathways in a quantum dot solid, highlighting the role of ligand engineering.
Quantitative Data: Ligand Impact on Device Performance
The theoretical principles of ligand engineering are validated by dramatic improvements in key device metrics. The following tables consolidate quantitative data from recent research, providing a clear comparison of how different ligand strategies impact performance.
Table 1: Ligand Impact on PbS Quantum Dot Solar Cell Performance
| Ligand Treatment | Device Architecture | Power Conversion Efficiency (PCE) | Key Parameters | Reference |
|---|---|---|---|---|
| TBAI (Inorganic) | ZnO/PbS-TBAI | 6.0 ± 0.4% | JSC = 20.7 ± 1.1 mA/cm², VOC = 0.506 V | [38] |
| TBAI + EDT (Bilayer) | ZnO/PbS-TBAI/PbS-EDT | 8.2 ± 0.6% (Certified: 8.55%) | JSC = 25.3 ± 1.1 mA/cm², VOC = 0.525 V | [38] |
| Ligand Customization | PbS CQD Solar Cell | 13.3% (vs. 10.4% control) | Improved passivation, suppressed band tail states | [39] |
Table 2: Ligand Impact on Electrical and Material Properties
| Ligand System | Material / Device | Key Measured Outcome | Implication for Charge Transport | Reference |
|---|---|---|---|---|
| n-butylamine | Nano-patterned PbS FET | Zero-bias conductivity: 17 µS/cm (180x increase vs. drop-cast) | Reduced structural defects and shorter tunneling distance | [16] |
| Ferrocene carboxylate | ZnO QD Film | Enabled long-range charge transport via redox ligand self-exchange | Introduced an active conduction pathway beyond simple tunneling | [4] |
| 3-mercaptopropionic acid | CdSe QD / MoS₂ Hybrid Phototransistor | Specific detectivity: 3.1 × 10¹³ Jones; ~100x faster response | p-doping effect maintained gate control, enhanced interfacial charge transfer | [40] |
| Carboxylic Acid | PbSe/PbS CQD FET | Carrier mobility increased 10-30x in ambient conditions over 72h | Initial surface passivation by H₂O/O₂ reduces trap states | [36] |
Experimental Protocols: Methodologies for Ligand Exchange
Reproducible device performance hinges on standardized, well-understood experimental procedures. The two primary ligand exchange techniques are detailed below.
Solid-State Ligand Exchange
This layer-by-layer method is widely used for fabricating QD thin films [36].
- Film Deposition: A substrate is spin-coated with a solution of QDs capped with long-chain native ligands (e.g., oleic acid) in a non-polar solvent, forming an as-cast film.
- Ligand Introduction: A polar solvent (e.g., acetonitrile) containing the new short-chain ligands (e.g., EDT, MPA, TBAI) is introduced onto the QD film.
- Soaking and Reaction: The film is soaked for an optimized duration (typically seconds to minutes) to allow the new ligands to replace the native ones on the QD surfaces.
- Purification: The film is spin-dried to remove the solution, followed by a washing step with a neat polar solvent to eliminate the displaced long ligands and any excess exchange agents.
- Layer Buildup: Steps 1-4 are repeated to build a film to the desired thickness.
Solution-Phase Ligand Exchange
This approach involves executing the ligand exchange while the QDs are still in solution, prior to film deposition, which can lead to superior uniformity and fewer defects [39] [16].
- Mixing: A solution of QDs with native ligands is mixed with a solution containing the new functional ligands.
- Precipitation and Cleaning: The QDs, now with exchanged ligands, are precipitated out of solution (often by adding an anti-solvent) and then centrifuged.
- Redispersion: The purified QD pellet is redispersed in a compatible solvent, ready for deposition as a high-quality, pre-functionalized film.
The workflow below visualizes the procedural hierarchy and key decision points in these fundamental ligand exchange protocols.
The Scientist's Toolkit: Essential Reagents and Materials
Successful experimentation in QD ligand engineering requires a suite of specialized reagents and materials. The following table catalogues key components and their functions.
Table 3: Research Reagent Solutions for Quantum Dot Ligand Engineering
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Lead Sulfide (PbS) QDs | Prototypical IR-active semiconductor QD; tunable bandgap. | Primary photoactive layer in solar cells and photodetectors [2] [38] [36]. |
| Tetrabutylammonium Iodide (TBAI) | Inorganic halide ligand for passivation and n-type character. | Creates the primary light-absorbing, charge-transporting layer in PbS CQD solar cells [38]. |
| 1,2-Ethanedithiol (EDT) | Short-chain, bidentate organic thiol ligand. | Used as an electron-blocking/hole-extracting layer in solar cell bilayers [38] [36]. |
| 3-Mercaptopropionic Acid (MPA) | Short-chain ligand inducing p-doping and surface dipoles. | Enhances photoresponse in hybrid MoS₂/QD phototransistors by modulating carrier concentration [40]. |
| Ferrocene Carboxylic Acid | Redox-active ligand for active charge transport pathways. | Enables self-exchange charge transport in ZnO QD assemblies, supplementing direct tunneling [4]. |
| n-Butylamine | Short organic amine ligand for enhanced interdot coupling. | Increases conductivity in nano-patterned PbS QD solids for transistor studies [16]. |
| MgZnO/ITO substrates | Common electron-accepting transparent electrode stack. | Serves as the substrate and cathode for QD solar cell devices [2] [38]. |
Emerging Trends and Future Outlook
The field of ligand engineering is rapidly advancing beyond simple conductive linkers.
- Ligand Customization for Defect Control: Emerging strategies decouple the roles of colloidal stabilization and property engineering by using ligand mixtures. This allows for independent optimization, leading to superior passivation, suppressed band tail states, and higher device performance [39].
- Understanding Fundamental Transport: The use of nano-patterning to create defect-free QD solids is revealing the intrinsic charge dynamics of percolation networks. This provides crucial insight for the rational design of QD solids with electronic properties that reflect a tunable, periodic potential, moving beyond disordered transport [41] [16].
- Probing Ultrafast Dynamics: Advanced techniques like Resonant Auger Spectroscopy (RAS) and Core-Hole Clock Spectroscopy (CHCS) are being used to map attosecond-scale electron transfer. This provides element-specific insights into how QD size and ligand environment influence the earliest charge delocalization steps, critical for long-range transport [2].
In conclusion, the deliberate tailoring of quantum dot surface ligands is a powerful and essential strategy for mastering charge transport. By carefully selecting ligand length, chemistry, and functionality, researchers can transform QD solids from poorly conducting assemblies into high-performance materials, paving the way for next-generation transistors, solar cells, and other advanced optoelectronic devices.
Overcoming Roadblocks: Managing Defects, Disorder, and Stability
Identifying and Passivating Surface Defects that Act as Non-Radiative Recombination Centers
In quantum dot (QD) solids, non-radiative recombination centers are defects, often on the QD surface, that cause electrons and holes to recombine without emitting light. This process significantly reduces the efficiency of optoelectronic devices such as solar cells and light-emitting diodes (LEDs) [42]. The surface chemistry of QDs, particularly the choice and coverage of surface ligands, is a critical factor in both the creation and passivation of these defects [43] [3]. This guide details the mechanisms of non-radiative recombination and provides a systematic, experimental approach to identifying and passivating these detrimental surface defects, with a specific focus on how surface ligand length modulates charge transport and recombination dynamics in QD solids.
Fundamentals of Non-Radiative Recombination
Mechanisms and Defect Types
Non-radiative recombination in semiconductors primarily occurs through the Shockley-Read-Hall (SRH) process, where defect states within the bandgap act as stepping stones, capturing electrons and holes sequentially via lattice vibrations (phonons) [42]. The textbook model assumes a single, mid-gap defect level is the most effective recombination center. However, first-principles studies reveal a more complex two-level process that can occur through relatively shallow levels [42]. In this mechanism:
- One type of carrier (e.g., an electron) is captured at a defect, forming a metastable state.
- The local defect configuration undergoes a rapid structural relaxation to a stable state.
- The other type of carrier (e.g., a hole) is then captured through a different energy level of the same defect, leading to recombination [42].
This two-level process can enhance recombination rates by orders of magnitude, meaning that defects not located near the mid-gap can still be potent recombination centers [42]. In QDs, the high surface-to-volume ratio makes surface atoms, which have unsaturated "dangling" bonds, predominant sources of these trap states.
Impact of Surface Ligands on Recombination
Surface ligands play a dual role: they passivate dangling bonds to reduce trap states, but they also dictate inter-dot coupling, which influences charge transport and energy transfer.
- Ligand Chain Length and Charge Transport: Shorter ligand chains reduce the interparticle distance, which exponentially increases the electron hopping rate between QDs [1] [4]. Electrochemical studies on InP/ZnSe/ZnS QDs demonstrate that shorter ligand chains (e.g., hexanoic acid vs. oleic acid) lower the energy barrier for charge injection, leading to higher current densities and more efficient charge transport in devices [44].
- Ligand Chain Length and Energy Transfer: Van der Waals interactions between long-chain ligands can force QDs into closer proximity, strengthening the Förster Resonance Energy Transfer (FRET) between dots. This inter-dot energy transfer can lead to energy-transfer-induced quenching, where energy migrates to a non-radiative trap site, reducing device efficiency and lifetime [44]. Controlling ligand length and coupling is therefore crucial to mitigate this loss pathway.
Experimental Identification and Characterization
A multi-faceted experimental approach is required to conclusively identify and quantify non-radiative surface defects.
Spectroscopic Techniques
Table 1: Spectroscopic Techniques for Identifying Non-Radiative Defects
| Technique | Measured Parameters | Information on Defects |
|---|---|---|
| Time-Resolved Photoluminescence (TRPL) | Photoluminescence decay lifetime | Surface defect density; distinguishes radiative vs. non-radiative recombination rates [45]. |
| Fluorescence Lifetime Imaging Microscopy (FLIM) | Spatial mapping of PL lifetime and intensity | Energy transfer population and efficiency between QDs; reveals heterogeneity influenced by ligand coupling [44]. |
| Transient Absorption (TA) Spectroscopy | Decay of excitonic bleach signal | Charge transfer dynamics from QD core to surface states; time constants increase with longer ligand chains [44]. |
Electrochemical and Electrical Probes
- Chronoamperometry: This technique measures current density over time in response to an applied potential. It directly probes charge injection efficiency into QDs. Studies show that decreasing ligand chain length from 18 to 6 carbon atoms can increase the integrated current density by a factor of 1.5, directly demonstrating the lower energy barrier for charge transport with shorter ligands [44].
- Cyclic Voltammetry (CV): CV acts as an electrochemical spectroscopy, providing a fingerprint of charge transfer processes in QD/ligand assemblies. It can identify the energetic positions of the QD's conduction band and any introduced redox ligand states, helping to map charge transport pathways [4].
Structural and Compositional Analysis
- X-ray Diffraction (XRD): XRD on QD solids can reveal how ligand chain length and the resulting interligand van der Waals coupling affect the interparticle spacing and structural order of the assembly [44].
- X-ray Photoelectron Spectroscopy (XPS): XPS, especially depth-profiling, confirms the successful binding and distribution of ligands across a QD film and can identify unpassivated surface atoms [4].
- Thermogravimetric Analysis (TGA): TGA quantitatively determines the ligand coverage on QD surfaces, which is a critical parameter for understanding passivation and transport [44].
Passivation Strategies via Surface Ligand Engineering
Ligand Selection and Classification
Ligands are categorized by their binding geometry and electronic function. Classically, L-type (Lewis base), X-type (anionic), and Z-type (Lewis acid, often metal carboxylate) ligands are used for passivation [1]. Effective passivation often requires careful management of Z-type ligands, which can passivate certain sites but also introduce mid-gap states if present in excess [1].
Table 2: Ligand Types and Their Impact on QD Properties
| Ligand Type | Examples | Primary Function | Effect on Charge Transport |
|---|---|---|---|
| Short-Chain Alkyl | Hexanoic Acid (HA), Decanoic Acid (DA) | Reduces inter-dot distance, improves charge injection [44]. | Hopping transport; high mobility [1]. |
| Conjugated Molecular | Arylamines, Thiophenes | Lowers tunneling barrier via molecular conjugation [1]. | Band-like transport; high mobility [1]. |
| Inorganic | Halides (I⁻, Cl⁻), Chalcogenides (S²⁻) | Strong passivation, minimal spatial footprint [43]. | Very high mobility due to tight dot coupling [43]. |
| Redox-Active | Ferrocene carboxylate (FcCOO⁻) | Introduces active electronic states for charge transport [4]. | Self-exchange chain reaction alongside electron hopping [4]. |
Advanced Ligand Engineering Strategies
- Atomic Ligand Passivation: This strategy uses monovalent halide anions (e.g., I⁻) to replace organic ligands. This approach provides excellent electronic passivation of surface defects while dramatically enhancing electronic transport due to the small size and strong binding of the atomic ligands [43].
- Redox Ligands as Active Transport Mediators: A paradigm shift from viewing ligands as passive spacers to using them as active components. For instance, ferrocene carboxylate ligands anchored to ZnO QDs provide a distinct pathway for charge transport via a self-exchange chain reaction between oxidized and reduced ferrocene units. This creates a complementary charge transport channel that operates in parallel with electron hopping through the QD conduction band [4].
- Defect-Tolerant Material Selection: Selecting intrinsic material systems that are less susceptible to defect formation is a foundational strategy. Emerging materials like Zintl-phase BaCd₂P₂ QDs show high defect tolerance, resulting in impressive photoluminescence quantum yields without complex surface treatments [46].
Experimental Protocols for Ligand Exchange and Analysis
Post-Synthetic Ligand Exchange on InP/ZnSe/ZnS QDs
This protocol outlines the replacement of native long-chain ligands with shorter carboxylic acids [44].
Materials:
- QD Source: As-synthesized InP/ZnSe/ZnS QDs capped with oleic acid (OA) from a single batch.
- Ligand Solutions: Decanoic acid (DA) and hexanoic acid (HA) in methanol.
- Solvents: Anhydrous hexane, methanol.
- Labware: Centrifuge tubes, benchtop centrifuge.
Procedure:
- Dissolve the native OA-capped QDs in an organic solvent like hexane.
- Add a molar excess of the new ligand (DA or HA) dissolved in methanol to the QD solution. Vigorous stirring is applied.
- Let the reaction proceed for a defined period (e.g., 1-2 hours). The QDs will precipitate out of solution.
- Isolate the QDs by centrifugation, discard the supernatant containing displaced OA, and redisperse the pellet in a fresh solvent.
- Repeat the purification steps (precipitation/redispersion) 2-3 times to remove residual OA and free ligands completely.
- Characterize the final product using TGA to determine new ligand coverage and NMR to confirm successful exchange and the absence of free ligands [44].
Electrochemical Characterization of Charge Injection
This protocol uses chronoamperometry to quantify how ligand length affects charge injection efficiency [44].
Materials:
- Working Electrode: Glassy carbon or ITO.
- Counter Electrode: Platinum wire.
- Reference Electrode: Ag/Ag⁺.
- Electrolyte: 0.1 M Tetrabutylammonium hexafluorophosphate (TBAPF₆) in acetonitrile.
- Samples: Solutions of OA-, DA-, and HA-capped InP/ZnSe/ZnS QDs in toluene.
Procedure:
- Prepare a standard three-electrode electrochemical cell with the QD solution.
- Apply a potential step from an initial potential (where no charging occurs) to a potential negative of the QD's conduction band edge (e.g., -1.7 V vs. Fc/Fc⁺).
- Record the current density as a function of time for several seconds.
- Integrate the current transient to obtain the total charge passed. A higher integrated charge density indicates more efficient charge injection into the QDs.
- Compare the integrated charge densities for QDs with different ligand lengths. The results will typically show that shorter ligands (HA) facilitate better charge injection than longer ones (OA) [44].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Surface Passivation Studies
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Oleic Acid (OA) | Long-chain native ligand; starting point for exchange [44]. | Provides colloidal stability but impedes charge transport. |
| Hexanoic Acid (HA) | Short-chain ligand for enhancing charge injection [44]. | Reduces inter-dot distance and energy barrier. |
| Tetrabutylammonium Iodide (TBAI) | Inorganic halide ligand source for atomic passivation [43]. | Significantly boosts mobility and passivates traps. |
| Ferrocene Carboxylic Acid | Precursor for redox-active ligands (FcCOO⁻) [4]. | Enables active self-exchange transport pathway. |
| 3-Mercaptopropionic Acid (MPA) | Short, bifunctional ligand (thiol binding group) [43]. | Can enhance photocatalytic performance in QD films [43]. |
| Zintl-Phase BaCd₂P₂ QDs | Defect-tolerant QD material system [46]. | Earth-abundant components; high initial PL without optimization. |
Effectively identifying and passivating non-radiative recombination centers is paramount for high-performance QD optoelectronics. A deep understanding of recombination mechanisms beyond the simple SRH model, combined with robust experimental characterization, is essential. The engineering of surface ligands—through strategic selection of their length, chemical structure, and electronic functionality—provides a powerful and versatile toolkit. By moving from passive spacers to active functional components, such as redox mediators, and by leveraging defect-tolerant materials, researchers can simultaneously suppress non-radiative recombination and engineer superior charge transport pathways in quantum dot solids.
In quantum dot (QD) solids, the precise control of energetic landscapes is paramount for advancing next-generation electronic and optoelectronic devices. Energetic disorder, which manifests as variations in energy levels within a QD ensemble, critically degrades charge transport and recombination dynamics. This technical guide examines two primary sources of this disorder: the size distribution of the quantum dots themselves and the incorporation of dopant ions. The influence of these factors is profoundly modulated by surface ligand chemistry, which dictates interdot coupling, determines the density of trap states, and ultimately controls charge carrier mobility in QD solids. A deep understanding of these interconnected variables provides the foundation for rationally designing high-performance QD-based materials.
The Fundamental Role of Surface Ligands in Energetic Landscapes
Surface ligands are not merely passive stabilizing agents; they are active components that directly define the electronic properties of QD solids. Their role is multifaceted, impacting both structural and electronic characteristics.
- Maintaining Individual Dot Properties: Ligands preserve the clean energy band gap of individual QDs within a solid film, preventing electronic coupling that could lead to disorder [3].
- Regulating Charge Transport: The length, functional groups, and surface coverage of ligands control charge carrier conduction across the solid film. Small chain organic ligands can be used to modulate mobility, dielectric constant, and carrier doping density [3].
- Providing Active Transport Pathways: Beyond acting as passive spacers, certain ligands can introduce electronic states that open additional charge transport paths. Redox-active ligands, for instance, enable a self-exchange chain reaction that facilitates long-range charge transport across the film [4].
- Reducing Energetic Disorder: Strategic ligand selection can minimize counterion-induced energetic disorder. Non-covalent interactions between ligands and polymer backbones (in doped systems) determine docking positions that significantly affect the resulting energetic disorder [47].
Ligand engineering thus serves as a critical strategy for flattening energy landscapes at QD-related interfaces, which is essential for achieving high-performance solution-processed photoelectronic devices [48].
Impact of Size Distribution on Energetic Disorder
Quantum confinement effects directly link a quantum dot's physical dimensions to its electronic structure. A heterogeneous size distribution within a QD solid inevitably creates a corresponding distribution of energy levels, constituting a significant source of energetic disorder.
Fundamental Mechanisms and Consequences
- Bandgap Variation: In QDs, the continuous energy bands of bulk materials split into discrete energy levels due to spatial carrier confinement. A key consequence is the increase in bandgap energy as QD size decreases [1]. A distribution of sizes therefore creates a distribution of bandgaps.
- Exciton Fine Structure: The exciton fine structure of QDs enables tailored degeneracy of electronic states at band edges. Inhomogeneous size distribution disrupts this fine structure, spreading hole populations among several states and increasing energy disorder between QDs and charge transport layers [48].
- Charge Transport Hindrance: Energetic disorder from size polydispersity creates energy barriers between neighboring QDs. Charge carriers require thermal activation to hop between dots of different sizes, reducing overall mobility and compromising device performance [1].
Experimental Approaches for Characterization and Mitigation
Advanced synthesis and characterization techniques are essential for quantifying and controlling size-dependent disorder.
Table 1: Experimental Techniques for Analyzing Size-Distribution-Induced Disorder
| Technique | Primary Function | Key Outcome Measures |
|---|---|---|
| Transmission Electron Microscopy (TEM) | Size and morphological analysis | Average size, size distribution, shape uniformity [48] |
| Absorption & Photoluminescence (PL) Spectroscopy | Optical property assessment | PL peak position, Full Width at Half Maximum (FWHM), Stokes shift [48] |
| Photo-assisted Kelvin Probe | Quasi-Fermi level splitting measurement | Contact Potential Difference (CPD), electron concentration [48] |
| X-ray Photoelectron Spectroscopy (XPS) | Surface chemistry and composition | Elemental composition, ligand binding efficiency [4] |
Synthesis Protocol: "Giant" Core/Shell QDs for Reduced Disorder
The synthesis of "giant" fully alloyed CdZnSe/ZnSeS core/shell QDs (size ~19 nm) demonstrates a strategic approach to mitigating size-related disorder [48]:
- Core Synthesis: Prepare CdZnSe alloy cores using hot-injection methods with metal precursors (e.g., cadmium acetate, zinc acetate) and selenium precursors (e.g., trioctylphosphine selenide) in high-temperature coordinating solvents.
- Shell Growth: Apply continuous ZnSeS shells using slow, layer-by-layer growth techniques with zinc oleate, selenium, and sulfur precursors to achieve smooth, compositionally graded interfaces.
- Structural Characterization: Employ HAADF-STEM element mapping and line scanning to verify homogeneous element distribution (particularly Cd and Se) and confirm delocalized hole wavefunctions.
- Optical Characterization: Measure absorption and PL spectra. The second derivative of absorption curves reveals ground-state band splitting, with alloyed QDs showing suppressed valence-band degeneracy and narrower FWHM (22 nm vs. 25 nm for conventional QDs), indicating reduced energetic disorder [48].
Figure 1: Experimental workflow for synthesizing and characterizing "giant" alloyed core/shell QDs with reduced energetic disorder [48].
Dopant-Induced Energetic Disorder and Control Strategies
Dopant ions introduce localized energy states that can either facilitate charge transport or create trapping sites, depending on their integration within the host material. The size and distribution of dopants critically influence the resulting energetic landscape.
Dopant Size Effects on Ionic Conduction
Research on lanthanide-doped ceria provides a paradigmatic example of how dopant size influences ionic conductivity and defect structures [49]:
- Optimal Dopant Size: Gd³⁺ (1.053 Å in 8-fold coordination) provides an optimal balance in doped ceria (GDC), exhibiting fewer defect clusters with vacancy pairs preferentially aligned along ⟨111⟩ and ⟨110⟩ directions. This configuration promotes a more open defect network that supports efficient oxygen-ion transport [49].
- Suboptimal Larger Dopants: The slightly larger Nd³⁺ (1.109 Å) in neodymium-doped ceria (NDC) promotes more compact defect configurations characterized by increased defect clustering and stabilized ⟨100⟩ vacancy alignment. This substantially reduces ionic conductivity despite the small difference in ionic radius [49].
- Cluster Formation: As dopant and vacancy concentrations increase, isolated defects tend to form more clustered configurations that hinder ion transport and suppress ionic conductivity [49].
Table 2: Impact of Dopant Size on Defect Structure and Conductivity in Doped Ceria
| Parameter | Ce₀.₈Gd₀.₂O₁.₉ (GDC) | Ce₀.₈Nd₀.₂O₁.₉ (NDC) |
|---|---|---|
| Ionic Radius | 1.053 Å (Gd³⁺) | 1.109 Å (Nd³⁺) |
| Preferred Vacancy Alignment | ⟨111⟩ and ⟨110⟩ | ⟨100⟩ |
| Defect Clustering Tendency | Lower | Higher |
| Defect Network Structure | More open | More compact |
| Relative Ionic Conductivity | 2-3 times higher | Lower |
Counterion Docking in Polymeric Semiconductors
In organic systems, the strategic positioning of dopant counterions presents a powerful method for reducing energetic disorder [47]:
- Computer-Aided Screening: A computational approach can screen optimal counterions by evaluating non-covalent interactions (NCIs) between counterions and polymers. This involves building 3D polymer packing structures, exploring potential docking sites, and calculating interaction energies for various counterions [47].
- NCI Composition Determination: The composition of NCIs (van der Waals vs. electrostatic interactions) determines counterion docking behavior. Cations with stronger electrostatic interactions (e.g., HPy⁺) dock closer to the polymer backbone, while those with dominant van der Waals interactions (e.g., MtBA⁺) diffuse into alkyl sidechain regions [47].
- Disorder Reduction: Optimal counterions that dock near the polymer backbone reduce conformational disorder, leading to narrower torsion angle distributions and extended orbital localization lengths at band edges, thereby enhancing charge transport [47].
Experimental Protocol: Counterion Docking for Reduced Disorder [47]
Computational Screening:
- Build three-dimensional polymer packing structure using molecular dynamics (MD) simulations.
- Identify potential docking sites within the alkyl sidechain region.
- Dock candidate counterions into refined binding pockets, generating multiple docking poses for each.
- Calculate interaction energies between polymer and counterions, focusing on non-covalent interaction composition.
Material Preparation:
- Dope the polymer system using standard molecular doping techniques.
- Implement ion-exchange method by immersing the doped polymer film in a solution containing the optimal counterion identified through screening.
- Rinse thoroughly to remove unexchanged ions and residual solution.
Characterization and Validation:
- Conduct MD simulations of polymer-counterion systems to verify docking positions.
- Perform energy decomposition analysis to quantify van der Waals and electrostatic interaction components.
- Analyze torsion angle distributions to assess backbone planarity improvement.
- Measure electrical conductivity and thermoelectric power factor to quantify performance enhancement.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Controlling Energetic Landscapes
| Reagent/Material | Function | Application Context |
|---|---|---|
| Short-Chain Organic Ligands | Modulate interdot distance, dielectric constant, and carrier doping density [3]. | QD solid films for photovoltaics and electronics |
| Redox Ligands (e.g., FcCOO⁻) | Provide active charge transport pathways via self-exchange reactions [4]. | Electrochemical devices, QD assemblies |
| Z-Type Ligands | Passivate surface traps, improving charge mobility [1]. | High-performance QD LEDs and solar cells |
| Lanthanide Dopants (e.g., Gd³⁺) | Create oxygen vacancies for ionic conduction with minimal defect clustering [49]. | Solid oxide fuel cell electrolytes |
| Aromatic Counterions (e.g., HPy⁺) | Dock near polymer backbones via electrostatic interactions, reducing conformational disorder [47]. | Doped polymeric semiconductors |
| "Giant" Alloyed QD Cores (CdZnSe) | Suppress valence-band degeneracy and ground-state band splitting [48]. | Low-voltage QLEDs with flattened energy landscapes |
Figure 2: Logical relationship between sources of energetic disorder, control strategies, and desired outcomes in quantum dot solids and doped materials.
The strategic control of energetic landscapes through management of size distribution and dopant effects represents a critical frontier in nanomaterials science. The precise synthesis of homogeneous, alloyed QD structures directly addresses size-dependent disorder, while rational dopant selection and placement—guided by an understanding of ionic interactions—mitigates dopant-induced disorder. Throughout this framework, surface ligand engineering serves as an essential cross-cutting strategy that actively modulates electronic coupling, passivates surface states, and in advanced implementations, creates entirely novel charge transport pathways. The continued refinement of these approaches, supported by the experimental protocols and characterization techniques outlined in this guide, promises to enable new generations of high-performance quantum dot and doped material devices with tailored electronic properties.
Strategies for Achieving Dense and Uniform Solid-State Packing of Quantum Dots
The assembly of colloidal quantum dots (QDs) into dense, uniform solid-state films represents a pivotal step in harnessing their exceptional optoelectronic properties for practical devices. While QDs in their colloidal state exhibit tunable bandgaps and high photoluminescence quantum yields, their performance in electronic and optoelectronic applications—including transistors, solar cells, and light-emitting diodes—is fundamentally governed by the quality of their solid-state packing [14]. Dense and uniform packing is paramount for efficient charge transport across the QD solid, a process critically dependent on the inter-dot distance and electronic coupling. This technical guide examines the principal strategies for achieving optimal QD packing, with particular emphasis on how surface ligand engineering dictates both structural integrity and charge transport functionality. The interplay between ligand chemistry, deposition techniques, and post-processing treatments creates a multi-dimensional design space for tailoring QD solids to specific application requirements, bridging the gap between nanocrystal synthesis and device-level performance.
Fundamental Principles: How Surface Ligands Govern Packing and Charge Transport
Surface ligands, organic molecules bound to the QD surface, play a dual and often contradictory role. They are essential for colloidal stability and size-tunable optical properties during synthesis, but in solid-state films, they become the primary mediator of both inter-dot spacing and electronic communication.
The Ligand Length Dilemma: Tunneling Barrier vs. Packing Density
The length of the surface ligand directly dictates the shortest possible distance between the inorganic cores of adjacent QDs. Longer, insulating ligands (e.g., oleic acid, oleylamine) create a significant potential barrier that suppresses charge transport by tunneling, while shorter ligands reduce this barrier but can challenge colloidal stability [4] [14]. This creates a fundamental trade-off: long ligands aid processing but hinder electronic coupling.
Beyond Passive Spacers: The Emergence of Redox-Active Ligands
A paradigm shift is underway, moving from viewing ligands as passive spacers to utilizing them as active components in the charge transport mechanism. Research demonstrates that ligands with electronic states, such as ferrocene carboxylate (FcCOO–), can create an additional pathway for charge transport via a self-exchange chain reaction, complementing traditional electron hopping through the QDs' conduction band [4]. This active ligand strategy represents a powerful method to enhance long-range charge transport without solely relying on ultra-short inter-dot distances.
Fabrication Techniques for Monolayers and Thin Films
The choice of fabrication method is critical for controlling the morphology, order, and scalability of QD solids. The following techniques are prominent in research and development.
Spin-Coating: Precision and Scalability
Spin-coating is a rapid, cost-effective, and scalable technique favored for producing uniform thin films. The process involves depositing a QD solution onto a substrate, which is then spun at high speed to spread the fluid via centrifugal force. Achieving a highly-ordered monolayer requires meticulous optimization of parameters [50].
Table 1: Key Parameters for Optimizing Spin-Coated QD Monolayers
| Parameter | Influence on Film Formation | Optimal Conditions for PbTe QDs [50] |
|---|---|---|
| QD Morphology | Determines packing geometry and defect density. | Spherical QDs (6–9 nm) achieve 90-100% coverage; cubical QDs (10–13 nm) achieve ~90% coverage. |
| Solvent Selection | Affects evaporation rate and dynamic wetting on the substrate. | Chloroform for cubical QDs; hexane for spherical QDs. |
| QD Concentration | Directly controls the density of deposited QDs. | Requires empirical tuning for each QD size and solvent system. |
| Substrate Properties | Surface energy and roughness influence QD adhesion and mobility. | TiO₂/ITO substrates successfully used for large-area (~3 cm²) monolayers. |
A study on PbTe QDs demonstrated that this method can produce a true monolayer (a layer with the height of a single QD) covering approximately 3 cm² with a root-mean-square roughness of just 1.37 nm [50]. The workflow for this optimized spin-coating process is systematic, as shown in the diagram below.
Layer-by-Layer (LbL) Deposition and Solid-State Ligand Exchange
For multilayer films, the layer-by-layer (LbL) method is highly effective. This iterative process involves depositing a single layer of QDs, followed by a solid-state ligand exchange to replace long native ligands with shorter conductive ones, and then repeating the cycle [4] [14]. This technique decouples the challenges of dense packing from the ligand exchange process, allowing for thicker films without compromising structural order or charge transport.
Surface Ligand Engineering for Enhanced Packing and Conductivity
Ligand exchange is the cornerstone of transforming an insulating QD solid into a conductive semiconductor.
Ligand Selection and Exchange Protocols
The choice of new ligand is critical. Shorter ligands like inorganic halides (e.g., I⁻, Br⁻), thiols, and carboxylates reduce the inter-dot distance, thereby increasing the electronic coupling strength. The exchange process can be performed in solution or in the solid state post-deposition. Solid-state exchange is often preferred as it preserves the pre-assembled QD superlattice [14]. The experimental protocol involves immersing the QD film in a solution containing the new ligand (e.g., a solution of ammonium iodide or ferrocene carboxylic acid in acetonitrile) for a controlled duration, followed by thorough rinsing to remove excess ligands and reaction by-products [4].
Impact of Ligand Functionality and Length
The functionality of the ligand directly influences the electronic properties of the QD solid. The diagram below illustrates the distinct charge transport pathways enabled by different ligand strategies.
Furthermore, even minor changes in ligand structure have measurable effects. A study on CdTe QDs demonstrated that the length of the surface ligand (specifically, 2-mercaptoacetic acid (TGA) versus 3-mercaptopropionic acid (MPA)) directly impacts the stability of the nanoparticle and its subsequent interactions at biological interfaces [51]. Although this study focused on toxicity, its finding that TGA (with a shorter carbon chain) forms a more stable complex with the CdTe core than MPA underscores a general principle: ligand structure dictates QD surface stability, which in turn influences the robustness of the solid-state film and its electronic properties.
Advanced Material and Processing Strategies
Purification and Precursor Control
Effective purification is an essential pre-processing step to remove excess ligands, unreacted precursors, and synthesis byproducts that can interfere with uniform packing. A prominent method is solid-phase extraction (SPE), which exploits the variable affinity of synthesis components towards a stationary phase. SPE is a facile, automatable alternative to conventional precipitation/redispersion cycles and can directly process crude reaction mixtures, improving preparation for deposition [52].
Molten Salt and Low-Temperature Synthesis
Innovative synthesis routes can also enhance final material properties. A low-temperature molten-salt method has been developed to synthesize carbon dots (CDs) with exceptionally high solid-state photoluminescence quantum yields (PL QYs) up to ~99.86% [53]. In this approach, a molten salt composed of NaCl, KCl, and ZnCl₂ acts as a reaction medium at mild temperatures (100–142 °C). The zinc ions coordinate with the forming CDs, suppressing the formation of non-radiative recombination channels and enhancing solid-state luminescence [53]. This principle of using an inorganic matrix to passivate surface states is highly relevant for preventing aggregation-caused quenching in closely packed QD films.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagent Solutions for QD Packing and Ligand Engineering Research
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Short Conductive Ligands | Solid-state ligand exchange to reduce inter-dot distance and enhance electronic coupling. | Inorganic halides (I⁻, Br⁻), Potassium hydroxide (KOH), Thiols, Carboxylates [4] [14]. |
| Redox-Active Ligands | Introduce active electronic states for a self-exchange charge transport pathway. | Ferrocene carboxylic acid (FcCOOH) [4]. |
| Optimized Solvents | Disperse QDs for deposition; choice affects evaporation rate and film uniformity. | Chloroform, Hexane, Toluene [50]. |
| Substrates | Support for QD film growth; surface properties critically influence monolayer formation. | ITO-coated glass, TiO₂/ITO [50] [4]. |
| Purification Materials | Stationary phase for solid-phase extraction (SPE) to purify QDs before deposition. | SPE columns/cartridges [52]. |
| Salt Precursors | For molten-salt assisted synthesis, enabling high-quality solid-state emitters. | NaCl, KCl, ZnCl₂ [53]. |
The pursuit of dense and uniform solid-state packing of quantum dots is a multi-faceted challenge that sits at the intersection of materials chemistry, process engineering, and device physics. The strategies outlined herein—from precision deposition techniques like optimized spin-coating to advanced ligand engineering with redox-active molecules—provide a robust toolkit for researchers. The critical insight is that surface ligands are not merely passive stabilizers but active design elements that control both the structural integrity of the QD solid and its electronic functionality. Future progress will likely involve the increased use of machine learning to optimize complex synthesis and deposition parameters [53], and the development of novel ligand chemistries that further minimize trade-offs between processability, stability, and charge transport. By systematically applying these strategies, the full potential of quantum dot solids in next-generation electronic and optoelectronic devices can be realized.
Mitigating Oxidation and Ensuring Long-Term Operational Stability of QD Films
The integration of quantum dots (QDs) into optoelectronic devices such as solar cells, light-emitting diodes (LEDs), and photodetectors necessitates their assembly into solid films where charge transport between individual QDs dictates device performance. A fundamental aspect of this charge transport is the surface chemistry of the QDs, specifically the length and chemical nature of the ligand species passivating their surfaces. While long, insulating native ligands are beneficial for colloidal stability, they severely impede inter-dot charge transport in films. Consequently, researchers employ ligand exchange to replace these with shorter organic or inorganic molecules. However, this process often exposes the QD core to environmental degradation, primarily through oxidation. Oxidation not only alters the optoelectronic properties of the QDs, leading to performance deterioration, but can also be intimately linked to the very charge transport pathways the ligand exchange sought to establish. This technical guide examines the mechanisms of QD film oxidation and outlines proven strategies to ensure their long-term operational stability, framed within the critical context of how surface ligand engineering affects charge transport in quantum dot solids.
Oxidation Mechanisms and Their Impact on Charge Transport
Understanding the degradation pathways is the first step toward developing effective mitigation strategies. For lead chalcogenide QDs like PbS and PbSe, the primary degradation mechanism is surface oxidation upon exposure to air and light.
- Chemical Transformation: X-ray photoelectron spectroscopy (XPS) studies reveal that air exposure of PbS QD thin films leads to the formation of lead sulfite (PbSO₃) and lead sulfate (PbSO₄) on the (100) facets of the QDs [54]. In some cases, the formation of lead oxide (PbO) and lead carbonate (PbCO₃) has also been observed [54].
- Consequences of Oxidation: This surface oxidation triggers a progressive blue-shift and quenching of the photoluminescence (PL) spectra [54]. The blue-shift occurs because oxidation effectively reduces the size of the semiconducting core, increasing the band gap—a direct manifestation of the quantum confinement effect. The PL quenching indicates the introduction of non-radiative trap states that compete with radiative recombination, thereby reducing the optoelectronic efficiency of the QD film.
- Link to Charge Transport: These chemical and physical changes directly impact charge transport. The formation of an oxidized shell can introduce insulating barriers between QDs, severely hampering electron hopping. Furthermore, the newly created trap states can localize charge carriers, reducing overall mobility and promoting recombination losses that diminish device performance.
Ligand-Dependent Oxidation Kinetics
Crucially, the rate and extent of oxidation are highly dependent on the surface ligands. Research demonstrates that this is a "ligand-dependent" process rather than an intrinsic property of the QDs [55]. For instance, while PbS QDs capped with 1,2-ethanedithiol (EDT) undergo significant oxidation, those coated with lead iodide/lead bromide (PbX₂) show remarkable resilience with no signs of oxidation or degradation under the same conditions [55]. This divergence underscores the pivotal role of ligand selection in determining device longevity.
The diagram below illustrates the ligand-dependent oxidation process and its consequences for charge transport.
Ligand Engineering Strategies for Enhanced Stability
Ligand engineering is the most potent tool for concurrently managing charge transport and stability. The strategic selection of ligands can passivate surface states, determine inter-dot coupling, and create a protective barrier against environmental factors.
Inorganic Ligands for Superior Passivation
Inorganic ligands, particularly halides, have emerged as the benchmark for achieving high performance and stability.
- Atomic Halides (I⁻, Br⁻): Ligand exchange with ions like iodide (I⁻) demonstrates complete removal of native organic ligands (e.g., oleylamine), as confirmed by the disappearance of C–H stretching bands in FTIR spectra [56]. This exchange results in dense, stable films. The key advantage is that these ligands passivate the QD surface without stripping cations (e.g., Pb²⁺), preventing the creation of defect states that act as traps for charge carriers [56].
- Lead Halides (PbX₂): Coating PbS QDs with a mixture of PbI₂ and PbBr₂ (PbX₂) provides exceptional stability. Studies show PbX₂-capped PbS QDs exhibit no signs of oxidation or degradation even in controlled oxygenated environments, whereas EDT-capped QDs from the same batch oxidize rapidly [55]. This robust passivation is crucial for maintaining consistent charge transport over time.
- Perovskite-like Shells (MAPbI₃): Treatment with methylammonium lead triiodide (MAPbI₃) can lead to the formation of a protective perovskite shell. While this shell may partially oxidize to PbO and PbCO₃, it effectively shields the core PbS QD, resulting in strong surface passivation and enhanced air stability [54].
The Role of Redox-Active Ligands
A shift from viewing ligands as passive spacers to active components in charge transport offers new stability avenues. Using redox ligands like ferrocene carboxylate (FcCOO⁻) introduces well-defined electronic states on the QD surface [4]. Charge transport in such assemblies can occur via two pathways:
- Electron hopping through the QDs' conduction band.
- Self-exchange through the immobile redox ligands [4].
This "active" transport pathway can potentially reduce reliance on perfect QD-to-QD contact, which may be compromised by minor surface oxidation, thereby offering a more resilient charge transport network.
Table 1: Comparison of Ligand Strategies for Stability and Charge Transport
| Ligand Type | Example | Impact on Stability | Impact on Charge Transport | Key Characteristics |
|---|---|---|---|---|
| Inorganic Halides | PbI₂, NH₄I | Excellent resistance to oxidation [55]. | High carrier mobility; n-type doping [54]. | Complete surface passivation; robust films. |
| Short-Chain Organic | EDT, MPA | Rapid oxidation, leading to performance evolution [55]. | Initially improves transport but degrades with oxidation. | p-type characteristic; ubiquitous but unstable. |
| Redox-Active | FcCOO⁻ | Potential for stable transport pathways. | Provides active self-exchange pathway alongside electron hopping [4]. | Introduces electronic states; Fermi-level tunable. |
| Perovskite Shell | MAPbI₃ | Good air stability; partial oxidation of shell [54]. | Conductive shell enhances inter-dot coupling. | Core-shell structure; strong surface passivation. |
Advanced Film Deposition and Device Engineering
Beyond ligand chemistry, the method of film deposition and overall device architecture play critical roles in mitigating degradation.
Single-Step Deposition from Pre-Exchanged Inks
Traditional layer-by-layer (LBL) solid-state ligand exchange is complex, can damage QD surfaces, and is unsuitable for thick films. Single-step deposition using QDs that have undergone in-solution ligand exchange prior to deposition is an advantageous alternative [56] [54]. This method yields denser, smoother films with less material waste and reduced surface degradation during fabrication [54]. Films deposited via this method from inks of PbS QDs treated with inorganic ligands like TBAI and MAPbI₃ show high stability, with minimal power conversion efficiency loss (under 1%) after 500 hours of storage in air [54].
Electrophoretic Deposition (EPD)
Electrophoretic deposition (EPD) is an efficient technique for assembling conformal, thick QD films, especially on non-planar substrates. Recent work has combined in-solution ligand exchange with EPD to directly deposit dense films of PbSe QDs capped with short NH₄I ligands [56]. This one-step process achieves fast deposition rates (1–100 nm/s) and produces films suitable for functional devices like IR photodetectors, bypassing the need for post-deposition, solid-state ligand exchanges that can compromise film integrity [56].
Charge-Balanced Device Architectures
Operational stability is not solely about chemical degradation; it also involves mitigating performance-degrading electrical processes. In QD-LEDs, a common issue is charge imbalance caused by excessive electron injection from a standard ZnO electron transport layer (ETL). This imbalance leads to efficiency droop and reduced operational lifetime [57].
A solution is the implementation of double metal oxide ETLs. Adding a layer of SnO₂ nanoparticles between the ZnO ETL and the QD layer significantly improves charge balance. The SnO₂ acts as a buffer, preventing spontaneous electron injection from ZnO due to its lower conduction band minimum, thereby protecting the QDs from high-energy charge flux and improving luminescence efficiency and stability [57].
The following diagram outlines a robust experimental workflow that incorporates these stability-enhancing strategies.
Experimental Protocols for Stability Assessment
To systematically evaluate the effectiveness of any stabilization strategy, researchers should implement standardized testing protocols.
Protocol: Air Exposure and Photo-Oxidation Stability Test
This protocol assesses the stability of QD thin films under ambient and light-soaked conditions [54].
- Sample Preparation: Prepare QD thin films on desired substrates (e.g., glass, FTO, ITO) using the deposition method under investigation (e.g., single-step spin-coating, EPD). Ensure a control sample is available.
- Baseline Characterization: Before air exposure, acquire:
- UV-vis-NIR Absorption Spectroscopy: To determine the initial band gap and quality of the film.
- Photoluminescence (PL) Spectroscopy: To measure the initial PL peak position and intensity.
- X-ray Photoelectron Spectroscopy (XPS): To establish the baseline surface chemical composition.
- Controlled Air Exposure: Place the samples in an ambient environment (controlled temperature and humidity, e.g., 25°C, 50% RH). For light-soaking tests, expose samples to continuous illumination from a solar simulator (e.g., AM1.5, 100 mW/cm²) in a controlled atmosphere chamber [55].
- Time-Resolved Monitoring: At regular intervals (e.g., 1h, 24h, 100h, 500h), repeat the PL and absorption measurements. Key indicators of oxidation are:
- Blue-shift of the absorption and PL peaks.
- Quenching of the PL intensity.
- Post-Analysis: After the test period, perform XPS again to identify the formation of oxidation products like PbSO₃, PbSO₄, or PbO [54] [55].
Protocol: Operational Stability of QD Solar Cells
This protocol evaluates the stability of complete devices under working conditions [55].
- Device Fabrication: Fabric QD solar cells with the structure: ITO/ZnO/PbX₂-PbS QD Active Layer/EDT-PbS QD Hole Layer/Au. For more stable devices, replace the EDT layer with a more robust alternative.
- Initial J-V Characterization: In a nitrogen glovebox, measure the current density-voltage (J-V) characteristics under simulated AM1.5 illumination to record the initial power conversion efficiency (PCE), fill factor (FF), short-circuit current (JSC), and open-circuit voltage (VOC).
- Environmental Degradation: Transfer devices to an environmental rig with controlled atmospheres (e.g., N₂, N₂ + 20% O₂, N₂ + 20% RH). Keep devices under continuous illumination at 100 mW cm⁻².
- Performance Tracking: Monitor the photovoltaic parameters (PCE, JSC, VOC, FF) in situ over time. A stable device will show a plateau in performance, while a degrading device will show a peak followed by a decline in these parameters [55].
Table 2: Key Reagents and Materials for Stable QD Film Research
| Reagent/Material | Function | Application Example |
|---|---|---|
| Lead Iodide (PbI₂) | Inorganic ligand for surface passivation and n-doping. | PbX₂-PbS active layer for high-efficiency, stable solar cells [55]. |
| Tetrabutylammonium Iodide (TBAI) | Inorganic ligand source for solution-phase exchange. | Creating stable, n-type PbS QD inks for single-step deposition [54]. |
| Tin Dioxide Nanoparticles (SnO₂ NPs) | Electron transport material with suitable band alignment. | Second ETL in ZnO/SnO₂ bilayer to suppress excessive electron injection [57]. |
| Ammonium Iodide (NH₄I) | Inorganic ligand for phase-transfer exchange. | Ligand exchange for PbSe QDs to create stable colloids in polar solvents for EPD [56]. |
| Methylammonium Lead Triiodide (MAPbI₃) | Perovskite ligand for core-shell passivation. | Surface treatment of PbS QDs to form a protective shell, enhancing air stability [54]. |
Achieving long-term operational stability in QD films is a multifaceted challenge that requires an integrated approach from the molecular to the device level. The central thesis, that surface ligand length and chemistry directly govern charge transport, must be expanded to include their paramount role in dictating oxidative stability. The transition from unstable short-chain organic ligands like EDT to robust inorganic passivants like PbI₂ represents the most significant step toward this goal. This must be complemented by advanced fabrication techniques like single-step deposition from pre-exchanged inks and device engineering strategies that manage internal charge balance. By adopting the ligand engineering principles, deposition methods, and standardized testing protocols outlined in this guide, researchers can systematically develop quantum dot solids that not only exhibit superior initial charge transport but also maintain their high performance throughout the operational lifespan of the device, thereby unlocking their full commercial potential.
The performance of quantum dot (QD) solids in optoelectronic devices is governed by a fundamental triad: efficient charge transport, superior optical properties, and long-term stability. At the heart of balancing these often-competing requirements lies the rational design of surface ligands. These organic or inorganic molecules cap the nanocrystal surface, playing a paradoxical role; they are essential for stabilizing colloidal QDs and preserving their optical properties, yet they can severely impede charge transport between adjacent dots [3] [1]. The length, chemical functionality, and binding group of these ligands directly control the interparticle spacing, electronic coupling, and trap state passivation, creating a complex trade-off that researchers must navigate [3] [4].
This technical guide details how surface ligand engineering dictates the electronic and optical properties of QD solids. It provides a structured framework for researchers to understand and manipulate the ligand-mediated trade-offs, supported by quantitative data, standardized experimental protocols, and visual workflows to inform the design of next-generation QD-based devices.
Fundamental Mechanisms: How Ligands Govern QD Behavior
Ligand-Dependent Charge Transport Pathways
Charge transport in QD solids occurs through several mechanisms, with the dominant pathway heavily influenced by surface ligand chemistry.
* electron Hopping: In this thermally activated process, charges "hop" from one QD to another. The ligand shell acts as a *tunneling barrier; the rate of hopping depends exponentially on the ligand chain length and the electronic coupling between dots [1]. Shorter ligands reduce the interparticle distance, thereby increasing the tunneling rate and overall charge mobility [4].
Band-Like Transport: In highly coupled QD solids, where inorganic cores are in close proximity, delocalized electronic states can form, allowing for band-like transport with higher mobility. Achieving this requires ultra-short or conductive ligands that minimize the potential barrier between cores [1].
Redox-Ligand Mediated Transport: A paradigm-shifting approach involves using electroactive ligands that provide an active pathway for charge transfer. For instance, ferrocene carboxylate ligands anchored to ZnO QDs create immobile redox states that enable a self-exchange chain reaction for long-range charge transport, complementing the hopping pathway [4].
Preserving Optical Properties and Stability
The primary functions of ligands that conflict with charge transport are surface passivation and colloidal stabilization.
Surface Passivation: A high surface-to-volume ratio makes QDs susceptible to surface defects that act as trap states for charge carriers, leading to non-radiative recombination and quenching of photoluminescence (PL). Effective ligands saturate these dangling bonds, preserving the photoluminescence quantum yield (PLQY) and the "clean" optical bandgap [3] [46].
Colloidal and Structural Stability: Ligands prevent QD aggregation in solutions and films. Furthermore, they protect the sensitive inorganic core from environmental degradation (e.g., oxidation). The choice of ligand headgroup and backbone is critical for maintaining the size, shape, and phase purity of QDs in solid films, which directly impacts device longevity [3] [1].
Quantitative Trade-Offs: A Data-Driven Perspective
The impact of ligand engineering can be quantified through key performance metrics. The table below summarizes the properties of different ligand classes, illustrating the inherent trade-offs.
Table 1: Impact of Ligand Class on QD Solid Properties
| Ligand Class | Typical Chain Length | Charge Mobility (cm²/V·s) | PLQY | Stability | Primary Trade-Off |
|---|---|---|---|---|---|
| Long Aliphatic (e.g., OA, OLA) | ~1-2 nm | Very Low (~10⁻⁷ to 10⁻⁵) | High | Good | Excellent passivation and stability at the cost of very poor conductivity. |
| Short/Inorganic Ligands | < 0.5 nm | Medium-High (~10⁻³ to 10⁻¹) | Variable | Moderate to Poor | High mobility can be achieved, but often at the cost of reduced stability and compromised passivation. |
| Conjugated Organic Ligands | ~1-1.5 nm | Medium (~10⁻⁴ to 10⁻²) | High | Good | Better conjugation enhances mobility while maintaining good optics, but synthesis can be complex. |
| Redox-Active Ligands | ~1 nm | Medium (with unique pathways) | Preserved | Good | Introduces an active transport pathway without requiring minimal spacing, but ligand design is complex [4]. |
The effect of ligand size on electronic properties is further quantified in specific studies. For instance, in lead sulfide (PbS) QD solids, systematic changes in the size and functional groups of small-chain organic ligands directly modulate the mobility, dielectric constant, and carrier doping density [3]. Furthermore, the size of the ligand itself can be used to tune the band gap of assembled solids, as seen in π-extended Carbo[n]Helicenes, where the band gap decreases from 3.83 eV to 3.32 eV as the molecular size and conjugation increase from [7] to [17] rings [58].
Table 2: Effect of Helicene Size on Electronic Properties [58]
| Helicene | HOMO-LUMO Gap (eV) | Reorganization Energy for Holes, λh (meV) | Reorganization Energy for Electrons, λe (meV) |
|---|---|---|---|
| [7]Helicene | 3.83 | 236 | 247 |
| [9]Helicene | 3.72 | 173 | 239 |
| [11]Helicene | 3.61 | 141 | 230 |
| [13]Helicene | 3.52 | 122 | 223 |
| [15]Helicene | 3.41 | 110 | 217 |
| [17]Helicene | 3.32 | 101 | 213 |
Experimental Protocols for Ligand Exchange and Characterization
Solid-State Ligand Exchange on PbS QD Films
This protocol is critical for fabricating conductive QD films for photovoltaics and photodetectors.
- QD Film Preparation: Spin-coat a film of PbS QDs capped with long-chain oleate ligands (e.g., from a hexane solution) onto a substrate. The film thickness is typically 50-100 nm.
- Ligand Solution Preparation: Prepare a 1-10 mM solution of the short target ligand (e.g., mercaptopropionic acid, MPA) in a polar solvent like methanol or acetonitrile. The choice of solvent is crucial—it must dissolve the new ligand but not swell or redissolve the QD film.
- Exchange Process: Drop-cast the ligand solution onto the PbS QD film and let it sit for 30-60 seconds. The short ligand diffuses into the film, displacing the native oleate ligands.
- Rinsing: Thoroughly rinse the film with the pure polar solvent to remove the displaced long-chain ligands and any excess exchange agent. This step is vital for film quality.
- Drying: Gently dry the film under a nitrogen or argon flow. The process can be repeated for multiple layers to build thicker, crack-free films with high conductivity [3].
Incorporating Redox-Active Ligands on ZnO QDs
This protocol details the functionalization of QDs with ligands that actively participate in charge transport.
- QD Synthesis and Film Casting: Synthesize ZnO QDs with native acetate/OH⁻ ligands. Drop-cast a film of these QDs onto an ITO substrate to a controlled thickness (e.g., 1.5 μm).
- Ligand Immersion: Immerse the ZnO QD film in a solution of the redox ligand, such as ferrocene carboxylic acid (FcCOOH), in acetonitrile overnight. This allows for complete ligand exchange.
- Rinsing and Validation: Rinse the film thoroughly with acetonitrile to remove any unbound FcCOOH. Successful anchoring is confirmed through a combination of techniques:
- XPS Depth-Profiling: To verify a uniform distribution of the redox species (e.g., Iron) throughout the film.
- NMR Spectroscopy: The absence of sharp peaks from free ligands in the solution used to rinse the film confirms no desorption and complete binding [4].
Visualization of Ligand Engineering Workflows
The following diagrams map the logical decision process for ligand selection and the experimental workflow for integrating and characterizing redox-active ligands.
Diagram 1: Ligand Selection Strategy for Target Application
Diagram 2: Workflow for Integrating Redox-Active Ligands
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents for Ligand Engineering Studies
| Reagent/Material | Function/Description | Application in Research |
|---|---|---|
| Oleic Acid (OA) / Oleylamine (OLA) | Long-chain aliphatic ligands used in synthesis for excellent passivation and colloidal stability. | Standard native ligands for QD synthesis; baseline for studying trade-offs against shorter ligands [3]. |
| Mercaptopropionic Acid (MPA) | A short-chain ligand with a thiol (-SH) binding group. | Common for solid-state ligand exchange to create conductive QD films for photovoltaics [3]. |
| Ferrocene Carboxylic Acid (FcCOOH) | A redox-active ligand providing electronic states for charge transport. | Used to create QD/redox ligand assemblies for studying self-exchange charge transport pathways [4]. |
| Tetrakis(dimethylamino)ethylene (TMEA) | Z-type ligand precursor (e.g., forms Cd(TMEA)₂). | Used for inorganic ligand exchange to achieve high carrier mobility and improved electronic coupling [1]. |
| Tris(2-carboxyethyl)phosphine (TCEP) | Biocompatible, water-soluble chalcogen transfer agent. | Enables sustainable, aqueous-phase synthesis of QDs, reducing reliance on toxic organic solvents [59]. |
| BaCd₂P₂ Zintl-Phase Precursors | Earth-abundant starting materials for defect-tolerant QDs. | Synthesis of QDs with intrinsic stability and high initial photoluminescence without complex passivation [46]. |
Emerging Strategies and Future Outlook
The field is moving beyond simple ligand exchange toward sophisticated multi-faceted strategies:
Ligand Bilayers and Graded Structures: A promising approach involves using a combination of ligands within a single device. For example, a long insulating ligand can be used on the surface of the QD film facing the environment to enhance stability, while the interface with the charge transport layer employs a short conductive ligand to facilitate charge extraction [1]. Computational models and machine learning are being leveraged to predict the properties of such complex systems and accelerate the discovery of optimal ligand combinations [60].
Exploitation of Intrinsic Material Properties: The discovery of new QD materials like Zintl-phase BaCd₂P₂ offers a pathway to reduce the dependency on perfect surface passivation. These materials exhibit defect tolerance, meaning they maintain high PLQY even with relatively unoptimized ligand coverage, thereby relaxing the stringent trade-off between optical properties and conductivity [46].
Sustainable and Scalable Production: Future research will increasingly integrate ligand design with green chemistry principles. The development of continuous flow processes in water using biocompatible chalcogen sources represents a significant step toward the responsible, large-scale production of functional QD solids [59]. The ultimate goal is a unified design framework where ligand chemistry, core composition, and assembly technique are co-optimized to deliver high-performance, stable, and commercially viable QD devices.
Probing Performance: Techniques for Validating Charge Transport Efficiency
Core-hole clock (CHC) spectroscopy has emerged as a powerful technique for probing ultrafast electron dynamics on attosecond timescales, providing unique insights into charge transfer processes that are fundamental to chemical reactivity and materials functionality. This technical guide explores the application of CHC spectroscopy for mapping attosecond charge transfer dynamics, with particular emphasis on its relevance to quantum dot solids research where surface ligand length critically determines charge transport efficiency. We present comprehensive experimental protocols, quantitative data analysis frameworks, and visualization tools that enable researchers to decipher electron delocalization mechanisms across diverse material systems. The integration of CHC spectroscopy with quantum dot research offers unprecedented opportunities for rationally designing advanced materials with tailored charge transfer properties for optoelectronic and energy applications.
Core-hole clock spectroscopy is an indirect time-resolved technique that utilizes the finite lifetime of a core-excited state as an intrinsic reference clock for measuring ultrafast electron dynamics [61] [62]. When an incident X-ray photon ejects or excites a core-level electron, it creates a highly unstable core-hole state that decays via Auger-Meitner or radiative processes with characteristic lifetimes typically ranging from hundreds of attoseconds to several femtoseconds [61]. This core-hole lifetime provides a natural temporal reference against which competing processes such as electron delocalization or charge transfer can be measured.
The fundamental principle underlying the CHC method involves comparing the rate of charge transfer to the rate of core-hole decay [62]. If an excited electron delocalizes from the atom on a timescale faster than the core-hole lifetime, the subsequent Auger-Meitner decay spectrum will reflect the altered electronic environment. By analyzing the spectral features in resonant Auger-Meitner or photoelectron spectra, researchers can extract upper limits for charge transfer times with attosecond resolution, surpassing the temporal limitations of conventional pump-probe techniques [61] [62].
Recent applications have demonstrated the particular utility of CHC spectroscopy for investigating charge-transfer-to-solvent (CTTS) states in aqueous systems [61] [63] and interfacial charge transfer in organic-inorganic hybrid materials [62]. The technique's ability to probe buried interfaces and complex environments without requiring ultrafast laser pulses makes it uniquely suited for studying quantum dot solids where surface chemistry governs electronic properties.
Fundamental Principles and Theoretical Framework
Quantum Mechanical Basis of the Core-Hole Clock Method
The CHC technique relies on the competition between two quantum processes: core-hole decay and electron delocalization. The core-hole lifetime (τ) follows an exponential decay profile described by e^(-t/τ), where τ is element-specific and varies with the core level involved [61]. For 1s core holes in light elements, lifetimes typically range from 1-10 femtoseconds, corresponding to attosecond temporal resolution [61].
When charge transfer occurs on a comparable timescale to core-hole decay, the resulting spectroscopic signature contains contributions from both the localized and delocalized electronic states. The branching ratio between these contributions provides a direct measure of the charge transfer rate (kCT) relative to the core-hole decay rate (kCH = 1/τ). The charge transfer time (τCT) can be estimated using the relationship:
τCT = τ × (IL/ID)
where IL and ID represent the intensities of spectral features corresponding to localized and delocalized states, respectively [61] [62].
Electronic Structure Considerations for Charge Transfer
The initial state in CHC spectroscopy involves a core-excited species with a localized electron wavepacket. For hydrated ions, this wavepacket rapidly evolves into charge-transfer-to-solvent (CTTS) states through scattering with neighboring solvent molecules [61]. The dynamics of this process depend critically on the energy of the excited electron relative to the ionization threshold and the electronic coupling between the donor and acceptor states.
In quantum dot systems, surface ligands modify this electronic coupling by acting as either passive tunneling barriers or active charge mediation sites [4]. The ligand's chemical structure, length, and electronic properties determine the height and width of the potential barrier between quantum dots, exponentially affecting charge transfer rates according to:
kCT ∝ e^(-βd)
where d is the ligand length and β is the attenuation factor dependent on the ligand's electronic structure [29].
Experimental Methodology and Protocols
Core-Hole Clock Spectroscopy Setup
Liquid-Jet Photoemission Spectroscopy: For investigating charge transfer in solution-phase systems, liquid-jet photoemission spectroscopy represents the state-of-the-art experimental approach [61]. The protocol involves:
Sample Preparation: Aqueous solutions of the target ions (e.g., Na+, Mg2+, Al3+) are prepared at concentrations typically between 0.1-1.0 M. High-purity salts and deionized water are essential to minimize contaminants [61].
Liquid Jet System: The solution is formed into a stable liquid jet (typically 10-30 μm diameter) in a vacuum chamber maintained at pressures below 10^-4 mbar. Microfluidic nozzles with precise temperature control (±0.1°C) ensure jet stability [61].
Photon Source: Synchrotron radiation facilities providing tunable X-ray energies (200-1500 eV) are required to access core levels. The photon energy is scanned across absorption edges to probe below, at, and above ionization thresholds [61] [63].
Electron Detection: Hemispherical electron analyers with acceptance angles of 30-60° and energy resolutions better than 100 meV are used to detect photoelectrons and Auger-Meitner electrons. Multichannel detection enables simultaneous measurement of multiple kinetic energies [61].
Data Collection Protocol:
- Measure Auger-Meitner spectra at multiple photon energies across the resonance
- Acquire reference spectra from gas-phase or solid standards for energy calibration
- Monitor sample purity through periodic survey scans
- Optimize counting statistics while minimizing radiation damage through dose-controlled measurements
Quantum Dot Film Preparation for Charge Transfer Studies
The investigation of ligand-dependent charge transport in quantum dot solids requires precise film fabrication protocols [29]:
Quantum Dot Synthesis: Lead sulfide (PbS) quantum dots with controlled sizes (3-8 nm diameter) are synthesized via hot-injection methods using oleic acid as capping ligands [29].
Ligand Exchange Procedure:
- Prepare 0.01-0.1 M solutions of short-chain ligands (e.g., 1,2-ethanedithiol [EDT], 1,3-benzenedithiol [1,3-BDT], mercaptocarboxylic acids [MPA]) in appropriate solvents [29]
- Deposit quantum dot films via layer-by-layer spin-coating (1000-3000 rpm for 30-60 s)
- Immerse films in ligand solution for 30-60 s followed by solvent rinsing
- Repeat for multiple layers to achieve desired film thickness (50-200 nm)
Film Characterization:
- Transmission electron microscopy (TEM) to determine inter-dot spacing
- X-ray photoelectron spectroscopy (XPS) to verify ligand binding and composition
- UV-Vis-NIR spectroscopy to monitor optical properties
- Photoluminescence spectroscopy to assess trap states [29]
Data Analysis and Interpretation Workflow
The analysis of CHC data involves a multi-step procedure to extract charge transfer times:
Spectral Decomposition: Auger-Meitner spectra are fitted to separate contributions from normal decay (localized electron) and spectator shift (delocalized electron) components [61].
Intensity Ratio Calculation: The relative intensities of localized (IL) and delocalized (ID) spectral features are determined through integration of fitted peaks.
Charge Transfer Time Calculation: Using the known core-hole lifetime (τ) and the intensity ratio, the charge transfer time is calculated as τCT = τ × (IL/ID) [61].
Energy Dependence Analysis: The procedure is repeated at multiple photon energies to map the dependence of charge transfer times on excitation energy [61].
Table 1: Core-Hole Lifetimes for Relevant Elements
| Element | Core Level | Lifetime (fs) | Applications |
|---|---|---|---|
| Sodium (Na) | 1s | ~1.7 | Hydrated ions [61] |
| Magnesium (Mg) | 1s | ~1.3 | Hydrated ions [61] |
| Aluminum (Al) | 1s | ~1.1 | Hydrated ions [61] |
| Sulfur (S) | 2p | ~1.4 | Quantum dot ligands [29] |
| Lead (Pb) | 4f | ~1.8 | Quantum dot cores [29] |
Key Experimental Findings and Data Analysis
Charge Transfer Dynamics in Aqueous Systems
Application of CHC spectroscopy to aqueous ions has revealed exceptionally fast electron delocalization timescales. Studies of isoelectronic Na+, Mg2+, and Al3+ ions demonstrate how ionic charge affects charge transfer dynamics [61]:
Table 2: Charge Transfer Times for Hydrated Ions via Core-Hole Clock Method
| Ion | Electronic Configuration | CTTS Formation Time (as) | Energy Dependence |
|---|---|---|---|
| Na+ | [Ne] | Several hundred as below threshold | Continuous decrease with photon energy [61] |
| Mg2+ | [Ne] | Intermediate values | Continuous decrease with photon energy [61] |
| Al3+ | [Ne] | As fast as 20 as far above threshold | Continuous decrease with photon energy [61] |
The continuous decrease in delocalization times with increasing photon energy indicates that excited electrons remain near the parent ion even above the ionization threshold, forming metal-ion electronic resonances associated with CTTS state manifolds [61]. These findings establish that the initial steps of electron hydration occur on attosecond timescales, significantly faster than subsequent solvent reorganization processes which occur on femtosecond timescales [61] [63].
Ligand-Engineered Charge Transport in Quantum Dot Solids
While direct CHC studies of quantum dots are limited in the current literature, extensive research on ligand effects provides crucial insights for designing CHC experiments. The relationship between ligand structure and charge transport properties has been systematically investigated [29]:
Table 3: Influence of Surface Ligands on Quantum Dot Film Properties
| Ligand Treatment | Inter-dot Distance (nm) | PL Peak Shift | Proposed Charge Transport Mechanism |
|---|---|---|---|
| Oleic Acid (OA) | 10.2 ± 0.8 | Reference | Tunneling through insulating barrier [29] |
| 1,3-BDT | 9.3 ± 0.8 | Pronounced red-shift | Conjugated pathway-mediated transport [29] |
| EDT | 7.8 ± 0.8 | Pronounced red-shift | Reduced tunneling barrier [29] |
| MPA | 7.6 ± 0.8 | Pronounced red-shift | Bidentate binding enhanced coupling [29] |
| (NH4)2S | 6.7 ± 0.7 | Pronounced red-shift | Inorganic sulfide bridges [29] |
The significant reduction in inter-dot spacing with shorter ligands correlates with enhanced electronic coupling between quantum dots. Photoluminescence red-shifts indicate the formation of delocalized states across multiple dots, suggesting the emergence of miniband transport in strongly coupled systems [29].
Advanced ligand design has evolved beyond passive spacers to incorporate active charge mediation functionalities. Redox-active ligands such as ferrocene carboxylate (FcCOO-) introduce electronic states that enable long-range charge transport via self-exchange chain reactions [4]. This represents a paradigm shift from modulating tunneling barriers to actively engineering charge transport pathways through molecular functionality.
Integration with Quantum Dot Research
Bridging CHC Spectroscopy and Quantum Dot Ligand Engineering
The integration of core-hole clock spectroscopy with quantum dot research offers exciting opportunities to directly probe ligand-mediated charge transfer dynamics with attosecond resolution. Specific research applications include:
Direct Measurement of Interdot Charge Transfer Times: CHC spectroscopy can quantitatively determine how ligand length and chemistry affect electron tunneling rates between quantum dots. By creating core holes on either the quantum dot core atoms or specific ligand elements, the technique can track electron movement across the ligand bridge [62].
Mapping Electronic Coupling Dependence: Systematic studies using quantum dot solids with controlled ligand architectures would enable precise determination of the distance dependence (β value) for charge transfer through different molecular frameworks. This provides fundamental parameters for designing optimal quantum dot solids [29] [4].
Active vs. Passive Ligand Roles: CHC spectroscopy can distinguish between charge transport through passive tunneling barriers versus active mediation by redox ligands. The technique could directly observe the superexchange mechanism through conjugated ligands or the hopping transport through redox-active centers [4].
Experimental Design for Quantum Dot CHC Studies
Future CHC investigations of quantum dot solids should incorporate these key design elements:
Sample Architecture: Fabricate well-defined quantum dot superlattices with controlled inter-dot spacing and coordination to minimize disorder effects [29].
Ligand Selection: Include both conventional insulating ligands (EDT, MPA) and advanced functional ligands (redox-active, conjugated) to compare charge transfer mechanisms [4].
Probe Selection: Utilize sulfur K-edge (2p core hole) for thiol-based ligands or lead M-edge for quantum dot core states to element-specific charge dynamics [29].
Complementary Techniques: Correlate CHC results with transient absorption spectroscopy, time-resolved photoluminescence, and electrical measurements to connect attosecond dynamics with device-relevant timescales [62].
Visualization of Core-Hole Clock Methodology
Diagram 1: Core-Hole Clock Spectroscopy Workflow. This flowchart illustrates the competitive processes between core-hole decay and charge transfer that form the basis of the CHC method.
Diagram 2: Energy Diagram of Core-Hole Clock Processes. This diagram shows the electronic states and transitions involved in CHC spectroscopy, highlighting the competition between core-hole decay and charge transfer.
Essential Research Reagent Solutions
Table 4: Key Research Reagents for Core-Hole Clock and Quantum Dot Studies
| Reagent Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Quantum Dot Cores | PbS, PbSe, ZnO, CdSe | Light-absorbing/heavy element for core-hole creation | Size uniformity critical for defined energy levels [29] |
| Passive Ligands | Oleic Acid, EDT, MPA, 1,3-BDT | Control inter-dot spacing and tunneling barriers | Chain length and binding affinity determine inter-dot distance [29] |
| Active Ligands | Ferrocene carboxylate, Tetrathiafulvalene derivatives | Provide redox states for hopping transport | Energy level alignment with quantum dot states crucial [4] |
| Aqueous Ions | Na+, Mg2+, Al3+ (isoelectronic series) | Model CTTS systems for method validation | Ionic strength affects hydration structure [61] |
| Synchrotron Targets | Silicon, gold, platinum | Reference materials for energy calibration | Clean, well-characterized surfaces required [61] |
Core-hole clock spectroscopy represents a powerful approach for mapping attosecond charge transfer dynamics in complex materials systems. The technique's unique ability to probe ultrafast electron delocalization without requiring femtosecond laser pulses makes it particularly valuable for investigating quantum dot solids where surface ligands dictate charge transport properties.
The integration of CHC spectroscopy with quantum dot research promises to address fundamental questions about how ligand structure and composition influence electron transfer mechanisms. By directly measuring charge transfer times through different molecular architectures, researchers can establish quantitative structure-property relationships to guide the rational design of quantum dot solids with enhanced charge transport characteristics.
Future advancements in CHC methodology will likely focus on extending the technique to more complex material systems, including multilayer quantum dot assemblies and operational device structures. The combination of CHC spectroscopy with complementary techniques such as attosecond transient absorption and time-resolved X-ray diffraction will provide a multidimensional view of charge transfer processes across temporal and spatial scales.
As quantum dot technologies continue to advance toward commercial applications in photovoltaics, lighting, and electronics, the insights gained from core-hole clock studies will play an increasingly important role in optimizing material performance through controlled interfacial design and tailored charge transfer pathways.
Electrochemical and Field-Effect Transistor (FET) Characterization of Carrier Mobility and Doping
The performance of quantum dot (QD) solids in electronic and optoelectronic devices is fundamentally governed by their charge transport properties, specifically carrier mobility and doping density. These properties are not intrinsic to the QDs alone but are extensively dictated by their surface chemistry. Within this framework, surface ligands act as critical tuning parameters, controlling interparticle spacing, electronic coupling, and surface passivation, thereby directly influencing charge transport mechanisms [3] [1]. This technical guide details the characterization of these essential parameters through Field-Effect Transistor (FET) and electrochemical methods, providing a standardized framework for evaluating how surface ligand length and functionality impact charge transport in QD solids.
Core Principles: Ligands and Charge Transport in QD Solids
In QD solids, charge transport occurs via tunneling or hopping between individual nanocrystals. The surface ligand shell, a monolayer of organic or inorganic molecules, forms the physical and electronic barrier through which charges must travel.
- Ligand Length: Shorter ligands reduce the interparticle distance, exponentially increasing the electron tunneling rate and enhancing carrier mobility [4] [1]. This is a primary strategy for improving conductivity.
- Ligand Functionality: The chemical nature of the ligand determines its role as a passive spacer or an active transport component. While insulating organic ligands act as passive barriers, redox-active ligands can introduce electronic states that provide a distinct, active pathway for long-range charge transport via self-exchange reactions [4].
- Surface Passivation: Effective ligands passivate surface trap states. Inadequate passivation leads to charge carrier trapping, which suppresses mobility and doping efficiency by immobilizing free carriers [3] [64].
The following diagram illustrates the primary charge transport pathways in a quantum dot solid, highlighting the role of both the QDs and the surface ligands.
Figure 1: Charge transport pathways in quantum dot solids. Carriers can tunnel between QD cores via short ligands or use the active pathway provided by redox ligands.
Field-Effect Transistor (FET) Characterization
The FET serves as a powerful tool for quantifying carrier mobility and doping type in QD solids. The QD film acts as the semiconductor channel, and its conductivity is modulated by a gate voltage.
Experimental Protocol for FET Fabrication and Measurement
Device Fabrication:
- Substrate Preparation: Use a heavily doped silicon wafer with a thermal oxide layer (typically 100-300 nm thick) as a global gate electrode and gate dielectric, respectively.
- Electrode Patterning: Define source and drain electrodes (often gold) onto the oxide surface using photolithography or shadow masking. The channel length (L) and width (W) are critical for mobility calculation.
- QD Film Deposition:
- Method A (Direct Deposition): Deposit a film of pre-synthesized, ligand-engineered QDs directly onto the substrate between the electrodes. This can be achieved via blade-coating or spin-coating of a stable colloidal ink [65].
- Method B (Layer-by-Layer): Alternatively, employ a layer-by-layer (LbL) spin-coating process, where each layer of QDs is subjected to a solid-state ligand exchange (e.g., dipping in a solution of short ligands) to remove long insulating ligands and promote electronic coupling [66] [65].
- Post-Deposition Treatment: A washing step (e.g., immersion in methanol) may be required to remove excess ligands and improve interparticle coupling, which has been shown to increase electron mobility by an order of magnitude [65].
Electrical Measurement:
- Setup: Use a semiconductor parameter analyzer in a probe station, preferably under vacuum or inert atmosphere to prevent device degradation.
- Transfer Curve Measurement: Sweep the gate voltage (VGS) while keeping the drain-source voltage (VDS) constant (e.g., at VDS = 0.1 V for the linear regime). Measure the resulting drain current (IDS).
- Output Curve Measurement: Sweep VDS at different fixed VGS values to observe IDS.
Data Analysis:
- Carrier Type: The direction of the gate voltage sweep that increases current indicates the majority carrier type (negative for n-type, positive for p-type).
- Carrier Mobility (μ): In the linear regime, the transconductance (gm = ∂IDS/∂VGS) is used to calculate mobility: μlin = (L / (W * Ci * VDS)) * gm where Ci is the capacitance per unit area of the gate dielectric.
- On/Off Ratio: The ratio of maximum to minimum IDS.
- Threshold Voltage (Vth): The gate voltage at which the channel begins to conduct, indicative of the doping density.
Impact of Ligands on FET Performance
The table below summarizes quantitative data from studies on how ligand engineering affects FET parameters in PbS QD solids.
Table 1: Effect of Ligand Exchange on FET Performance of PbS QD Solids
| Ligand Type | Deposition Method | Mobility (cm²/V·s) | On/Off Ratio | Key Finding | Source |
|---|---|---|---|---|---|
| Long Organic (Oleate) | Spin-coating (LbL) | < 10⁻⁴ (Est.) | Low | Insulating behavior due to long tunneling distance | [65] |
| Inorganic Iodide (I⁻) | Blade-coating (Single Step) | ~0.12 (after washing) | 10⁶ | Demonstrates high performance with scalable method | [65] |
| Inorganic Halometallate (PbI₃⁻) | Blade-coating (Single Step) | Higher than I⁻ counterpart | - | Improved passivation and transport over simple halides | [65] |
| Optimized Short Ligands | Spin-coating (LbL) | > 30 | - | Highlights potential of proper ligand choice & treatment | [66] |
Electrochemical Characterization
Electrochemical methods provide a powerful means to probe the density of states, doping levels, and charge transport kinetics in QD solids, often in situ.
Experimental Protocol for Cyclic Voltammetry (CV)
Cell Assembly:
- Working Electrode: A conductive substrate (e.g., ITO, FTO, or gold) coated with the film of ligand-capped QDs.
- Counter Electrode: An inert wire, such as platinum.
- Reference Electrode: A stable reference like Ag/AgCl or a quasi-reference. All potentials are often reported versus the Fc/Fc⁺ (ferrocene/ferrocenium) couple for internal consistency.
- Electrolyte: A 0.1 M solution of a supporting electrolyte (e.g., TBAPF₆) in a dry, deaerated organic solvent (e.g., acetonitrile or dichloromethane).
Measurement:
- Record a cyclic voltammogram by sweeping the potential applied to the working electrode across a defined range (e.g., from -0.6 V to +0.9 V vs. Fc/Fc⁺) and measuring the current.
- The scan typically reveals distinct signals: a broad wave corresponding to charge injection into the QD's conduction band and sharp, quasi-reversible peaks corresponding to the redox activity of surface-bound ligands (if present) [4].
Data Analysis:
- Doping Density: The charge (Q) passed during the reduction or oxidation of the QD film can be integrated and used to calculate the carrier density using the formula: N = Q / (e * V), where e is the elementary charge and V is the film volume.
- Energetics: The onset potentials for QD charging provide information about the conduction and valence band edge positions.
- Ligand Redox States: The position and reversibility of peaks from redox-active ligands (e.g., ferrocene carboxylate) confirm their successful anchoring and electronic communication with the QD core [4].
Probing Active Transport via Redox Ligands
Electrochemistry is uniquely suited to characterize systems with redox-active ligands. In such assemblies, charge transport can occur via two parallel pathways:
- Electron hopping through the QD conduction band.
- Self-exchange through the immobile redox ligands, an active pathway enabled by the ligand's electronic states [4].
The following workflow diagram outlines the key steps for fabricating and characterizing a QD solid with redox-active ligands.
Figure 2: Workflow for characterizing charge transport in redox-ligand functionalized QD solids.
The table below compares the key characteristics of these two charge transport pathways as identified through electrochemical studies.
Table 2: Charge Transport Pathways in QD/Redox Ligand Assemblies
| Characteristic | Pathway 1: QD Band Hopping | Pathway 2: Ligand Self-Exchange |
|---|---|---|
| Mechanism | Electron hopping/tunneling between QD cores | Self-exchange chain reaction between immobilized redox ligands |
| Dependence on Ligand Coverage | Weak (primarily dependent on inter-dot distance) | Strong, follows percolation theory |
| Kinetics | Faster, independent of ion transport | Slower, requires ion transport for charge compensation |
| Energetic Position | Governed by QD conduction band edge | Governed by ligand redox potential (e.g., +0.1 V vs. Fc/Fc⁺ for FcCOO⁻) |
| Role of Ligands | Passive spacers | Active electronic components |
The Scientist's Toolkit: Essential Research Reagents
This section catalogs key reagents and materials essential for experiments in this field.
Table 3: Essential Reagents for QD Charge Transport Studies
| Reagent/Material | Function/Description | Example Use Case |
|---|---|---|
| Lead Sulfide (PbS) QDs | Prototypical semiconductor QD with tunable IR absorption and emission. | Active layer in high-performance FETs and photovoltaics [3] [65]. |
| Cesium Lead Bromide (CsPbBr₃) Perovskite NCs | Emerging perovskite material with high photoluminescence quantum yield. | Studying the effect of ligands on surface states and non-radiative recombination [64]. |
| Oleic Acid / Oleylamine | Common long-chain organic ligands used in colloidal synthesis. | Provide initial colloidal stability; replaced via exchange for device fabrication [64] [65]. |
| Methylammonium Iodide (MAI) | Small inorganic ligand source for halide passivation. | Phase-transfer ligand exchange to create conductive PbS QD inks [65]. |
| Ferrocene Carboxylic Acid (FcCOOH) | Source of redox-active ferrocene carboxylate ligand. | Functionalizing ZnO QDs to create an active charge transport pathway [4]. |
| Propylene Carbonate | High-dielectric-constant, polar aprotic solvent. | Dispersing medium for inorganic-capped QD inks [65]. |
| Tetrabutylammonium Hexafluorophosphate (TBAPF₆) | Electrolyte salt for electrochemical cells. | Supporting electrolyte in non-aqueous electrochemistry [4]. |
Integrated Data Interpretation and Correlation
A comprehensive analysis requires correlating data from FET and electrochemical measurements. For instance, a positive shift in the FET threshold voltage after a specific ligand exchange, as observed in studies [65], indicates a reduction in fixed negative charge or an increase in hole doping within the film. This can be cross-referenced with CV data, which may show a new oxidation wave corresponding to the incorporated ligand's redox activity [4]. Furthermore, a significant increase in mobility from FET measurements, coupled with a change in the Stokes shift and PL lifetime observed in optical studies [64] [65], confirms that the new ligand regimen has successfully improved electronic coupling and passivated trap states. This multi-faceted approach is critical for unequivocally establishing the structure-property relationships that govern charge transport in ligand-engineered QD solids.
Quantum dot (QD) solids have emerged as a revolutionary class of materials for next-generation optoelectronic devices, including photovoltaics, light-emitting diodes (LEDs), and photodetectors. The performance of these devices hinges critically on charge transport through the QD solid, a process profoundly influenced by surface chemistry. Surface ligands—molecules bound to the QD surface—dictate inter-dot spacing, electronic coupling, and trap state passivation, thereby governing charge mobility. This review provides a comparative analysis of charge transport mechanisms in lead sulfide (PbS), indium phosphide (InP), and perovskite QD solids, framed within the broader thesis that surface ligand engineering is a universal strategy for enhancing device performance. We examine how ligand chemical structure and length modulate electronic properties across these material systems, synthesizing recent experimental findings to guide future research and development.
Fundamental Mechanisms of Charge Transport in QD Solids
Charge transport in quantum dot solids occurs primarily via quantum tunneling and thermally activated hopping between neighboring nanocrystals, as the long organic ligands used in synthesis create potential barriers that disrupt band-like transport [1]. The electronic coupling between dots, and thus the carrier mobility, is exponentially dependent on the inter-dot distance and the height of the tunneling barrier [29] [1].
- Inter-dot Distance: Shorter ligand molecules reduce the separation between quantum dot cores, facilitating stronger electronic coupling and more efficient charge carrier tunneling.
- Tunneling Barrier: The chemical nature of the ligand (e.g., its electronic structure) influences the energy barrier that carriers must overcome to hop between dots.
- Surface Passivation: Effective ligands passivate dangling bonds on the QD surface, reducing the density of trap states that cause non-radiative recombination and impede charge transport.
The competition between radiative recombination and non-radiative trap-assisted recombination directly impacts the efficiency of optoelectronic devices. Consequently, ligand engineering serves the dual purpose of enhancing charge transport while minimizing detrimental recombination pathways.
Comparative Analysis of QD Material Systems
PbS (Lead Sulfide) Quantum Dots
PbS QDs possess a large Bohr exciton radius (~18 nm), enabling strong quantum confinement effects and widely tunable optical properties across the infrared spectrum [2]. Research has consistently demonstrated that short-chain ligands dramatically enhance charge transport in PbS QD solids.
Key Findings:
- Ligand-Dependent Inter-Dot Spacing: A study comparing 1,2-ethanedithiol (EDT), 1,3-benzenedithiol (1,3-BDT), mercaptocarboxylic acids (MPA), and ammonium sulfide ((NH₄)₂S) found that these ligands reduced the inter-dot separation from the original oleic acid (OA)-capped state by 2.4 nm, 0.9 nm, 2.6 nm, and 3.5 nm, respectively. The smallest spacing from (NH₄)₂S treatment, which creates an inorganic, sulfide-rich interconnection between dots, highlights the ultimate limit of this approach [29].
- Enhanced Photovoltaic Performance: The incorporation of a PbS-EDT hole transport layer (HTL) in PbS-I CQD solar cells increased the power conversion efficiency (PCE) from 1.8% (without HTL) to 10.8%. This improvement was attributed to minimized nonradiative carrier recombination and improved charge extraction [67].
- Novel Perovskite-like Ligands: Recent work introduced a robust 2D perovskite-like ligand, (BA)₂PbI₄ (BA = butylammonium), for surface engineering. This ligand forms a thin shell on the QD surface, effectively passivating challenging non-polar facets. Solar cells using (BA)₂PbI₄-capped large-bandgap PbS CQDs achieved a PCE of 8.65%, while small-bandgap CQDs reached 13.1%, outperforming PbI₂-capped controls (11.3%) and demonstrating enhanced ambient stability [68].
- Ultrafast Charge Transfer: Resonant Auger Spectroscopy studies revealed that charge transfer times in PbS QDs are influenced by their size and ligand environment. Larger PbS QDs exhibited faster charge transfer rates compared to smaller ones, and the ligand shell (e.g., PbI₂) plays a critical role in determining these dynamics [2].
Table 1: Impact of Different Ligands on PbS QD Solid Properties
| Ligand | Ligand Type | Inter-Dot Spacing | Key Effects on Charge Transport | Device Performance (PCE) |
|---|---|---|---|---|
| Oleic Acid (OA) | Long-chain (C18) | Large (Reference) | Insulating barrier, poor transport | N/A |
| 1,2-ethanedithiol (EDT) | Short-chain dithiol | Reduced by 2.4 nm | Improved hopping, used as HTL | 10.8% (with HTL) [67] |
| (NH₄)₂S | Inorganic | Reduced by 3.5 nm | Inorganic coupling, minimal spacing | N/A [29] |
| PbI₂ | Metal Halide | Short | Good passivation, common reference | 11.3% (Control) [68] |
| (BA)₂PbI₄ | 2D Perovskite | Short | Facet-specific passivation, stability | 13.1% (Champion) [68] |
InP (Indium Phosphide) Quantum Dots
As a less toxic alternative to Cd- and Pb-based QDs, InP has gained significant attention. Similar to PbS, its charge transport properties are strongly modulated by surface ligand chemistry.
Key Findings:
- Ligand Chain Length and Charge Injection: Electrochemical studies on InP/ZnSe/ZnS core/shell/shell QDs with oleic acid (OA, C18), decanoic acid (DA, C10), and hexanoic acid (HA, C6) ligands demonstrated that shorter alkyl chains facilitate more efficient charge injection. Chronoamperometry measurements showed that decreasing the carbon atom count in the ligand from 18 to 6 increased the integrated current density by a factor of 1.5, indicating a lower energy barrier for charge transport [44].
- Inter-QD Energy Transfer: The same study used fluorescence lifetime imaging microscopy (FLIM) to reveal that longer ligand chains increase the population and efficiency of Förster Resonance Energy Transfer (FRET) between neighboring QDs. This energy transfer can lead to nonradiative recombination losses, thereby reducing the efficiency and lifetime of QD-LEDs. Shorter ligands weakened interligand van der Waals interactions, reducing FRET and its detrimental effects [44].
Table 2: Impact of Ligand Chain Length on InP/ZnSe/ZnS QD Properties
| Ligand | Chain Length (C#) | Charge Injection Efficiency | FRET Efficiency | Implication for QD-LEDs |
|---|---|---|---|---|
| Oleic Acid (OA) | C18 | Low (Reference) | High | Increased non-radiative loss, shorter lifetime |
| Decanoic Acid (DA) | C10 | Moderate | Moderate | Balanced properties |
| Hexanoic Acid (HA) | C6 | High (1.5x integrated current) | Low | Improved brightness and operational lifetime |
Perovskite Quantum Dots
Perovskite QDs, such as CH₃NH₃PbI₃ (methylammonium lead iodide), combine the defect tolerance of perovskites with the quantum confinement of nanocrystals. Ligand engineering in these systems often focuses on integrating the perovskite material itself as a functional ligand.
Key Findings:
- CH₃NH₃PbI₃ as a Multifunctional Ligand: A solid-state ligand exchange method was used to cap PbS QDs with CH₃NH₃PbI₃, replacing the original oleate/oleylamine ligands. This strategy offered several advantages: (1) it broadened the absorption spectrum to include visible light; (2) it enabled the fabrication of relatively thick, smooth active-layer films; and (3) it created a cascade energy alignment with the electron-transport layer (TiO₂) to facilitate charge transport. Solar cells utilizing these materials achieved a PCE of 4.25% with a high short-circuit current density (Jₛc) of 24.83 mA cm⁻² [69].
- Versatility of 2D Perovskite Ligands: As discussed in the PbS section, the 2D perovskite (BA)₂PbI₄ has also been successfully applied as a ligand, demonstrating that perovskite-like ligands are a versatile strategy applicable to different QD systems [68].
Experimental Methodologies and Protocols
A deep understanding of charge transport requires sophisticated characterization techniques. Below are detailed protocols for key experiments cited in this review.
Objective: To quantitatively evaluate the efficiency of charge injection into QDs with different surface ligands in solution.
- Procedure:
- Prepare solutions of QDs (e.g., InP/ZnSe/ZnS) passivated with the ligands under study (e.g., OA, DA, HA) in a suitable solvent.
- Use a standard three-electrode electrochemical cell: QD solution as the working electrode, a platinum counter electrode, and a reference electrode (e.g., Ag/AgCl).
- Apply a potential step (a voltage pulse) to the working electrode, driving it to a value sufficient to inject charge carriers (electrons or holes) into the QDs.
- Monitor the current density transient as a function of time.
- Integrate the current transient over time to obtain the total charge passed. A higher integrated current density indicates more effective charge injection into the QDs.
- Data Interpretation: The current density is dependent on the concentration of charged dots. A higher current density at the onset potential for a given ligand system signifies a lower energy barrier for charge transport.
Objective: To spatially map and quantify energy transfer (FRET) dynamics between QDs in a solid film.
- Procedure:
- Fabricate highly ordered solid films of the ligand-exchanged QDs on a substrate.
- Place the sample under a microscope equipped with a FLIM system.
- Excite the sample with a pulsed laser (e.g., 470 nm, 10 MHz repetition rate) focused through a high-resolution objective (e.g., 100x oil-immersion).
- For each pixel in a 2D scan (e.g., 512x512 pixels over a 5x5 µm² area), record the photoluminescence (PL) decay curve using time-correlated single-photon counting (TCSPC).
- Analyze the PL decay curves to extract the lifetime (τ) at each pixel and create a false-color lifetime map of the sample.
- Data Interpretation: A shorter average PL lifetime in the film compared to isolated QDs, or the presence of pixels with very short lifetimes, indicates efficient nonradiative energy transfer (FRET) to neighboring dots or trap states. Comparing lifetime maps for different ligands reveals how ligand chain length tunes inter-QD coupling.
Objective: To probe ultrafast (attosecond to femtosecond) charge transfer dynamics at the Pb sites in PbS QDs with site-specificity.
- Procedure:
- Prepare solid films of PbS QDs of varying sizes and ligand environments (e.g., PbI₂-passivated) on appropriate substrates.
- At a synchrotron beamline, expose the sample to tunable hard X-rays and scan the photon energy across the Pb M-edge (a core-level absorption edge).
- For each incident photon energy, measure the kinetic energy of the emitted Auger electrons (Pb MNN region).
- Construct a 2D resonant Auger map (intensity vs. incident photon energy and kinetic energy of emitted electrons).
- Analyze the map to distinguish between two decay channels: a) Localized (Raman) decay: The excited electron remains on the Pb atom, and b) Delocalized (Auger) decay: The excited electron has transferred away from the Pb atom within the core-hole lifetime (~0.26 fs for Pb 3d).
- Data Interpretation: The intensity ratio between the delocalized and localized decay channels provides an estimated charge transfer time. Faster transfer times indicate more efficient initial charge delocalization, a crucial step for long-range transport in devices.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Ligand Engineering and QD Solid Fabrication
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| Oleic Acid (OA) | Long-chain native ligand for QD synthesis | Provides colloidal stability; must be exchanged for efficient transport [44] [29] |
| 1,2-ethanedithiol (EDT) | Short-chain ligand for exchange | Reduces inter-dot spacing; improves charge hopping; used as HTL [67] [29] |
| Lead Iodide (PbI₂) | Inorganic passivating ligand | Common for PbS QDs; provides good passivation and short inter-dot distance [2] [68] |
| (BA)₂PbI₄ Precursor | 2D perovskite-like ligand | Enables facet-specific passivation, enhances stability and PCE [68] |
| CH₃NH₃PbI₃ Precursor | Perovskite matrix/ligand | Multifunctional ligand; broadens absorption, improves film formation [69] |
| Ammonium Sulfide ((NH₄)₂S) | Inorganic ligand | Creates metal-sulfide bonds between QDs; minimizes inter-dot spacing [29] |
| Mercaptocarboxylic Acids (e.g., MPA) | Short, bifunctional ligands | Can bind via thiol and carboxylate groups; reduces inter-dot distance [29] |
Ligand Engineering Workflow and Charge Transport Pathways
The following diagram illustrates the general workflow for ligand engineering and its direct impact on the charge transport pathways within a QD solid.
This comparative analysis unequivocally demonstrates that surface ligand engineering is a central, universal strategy for controlling charge transport in quantum dot solids, irrespective of the core material composition. While PbS, InP, and perovskite QDs each possess unique optoelectronic properties, they all adhere to the fundamental principle that shorter ligand chains enhance inter-dot electronic coupling and charge carrier mobility. The emergence of multifunctional ligands, particularly halide perovskites, represents a significant advancement beyond simple spacers, offering simultaneous passivation, enhanced absorption, and improved stability. Future research should focus on developing novel ligand chemistries that provide atomic-level precision in passivation, further reduce transport barriers, and enhance the long-term operational stability of QD solids under real-world conditions. The insights and protocols outlined in this review provide a foundational toolkit for researchers aiming to harness the full potential of quantum dot materials in advanced energy and electronic devices.
This technical guide explores the fundamental relationship between surface ligand chemistry, charge transport dynamics in quantum dot (QD) solids, and macroscopic device performance metrics, specifically power conversion efficiency (PCE) and external quantum efficiency (EQE). Surface ligands are not merely passive stabilizers but active components that dictate electronic coupling, trap state passivation, and charge transport mechanisms in QD assemblies. By examining recent advances in ligand engineering strategies—including short-chain ligands, conjugated molecules, and redox-active ligands—this review provides a comprehensive framework for linking molecular-scale surface modifications to enhanced device performance across photovoltaic and optoelectronic applications.
Quantum dot solids represent an emerging class of semiconductors where precisely engineered nanocrystals self-assemble into functional materials. The electronic coupling between individual QDs in these solids primarily occurs through their surface ligands, making ligand chemistry a critical determinant of device performance. Surface ligands originally serve to stabilize colloidal QDs during synthesis but become pivotal in mediating interdot charge transport in solid-state films [3]. Ligand engineering directly influences key performance metrics including PCE in solar cells and EQE in light-emitting devices by modulating conductivity, mobility, and recombination dynamics [70].
The fundamental challenge stems from a critical trade-off: long-chain native ligands (e.g., oleic acid, oleylamine) provide excellent colloidal stability but impede charge transport by acting as insulating barriers between QDs. Consequently, developing strategic ligand exchange processes has become a central focus in QD research to achieve both stability and efficient charge transport [1]. This review systematically examines how strategic ligand design at the microscopic level translates to enhanced macroscopic device performance.
Fundamental Charge Transport Mechanisms in QD Solids
Understanding how charges move through QD solids is essential for rationally designing high-performance devices. Charge transport in these assemblies occurs primarily through two distinct mechanisms, with ligand chemistry determining the dominant pathway.
Polaron Hopping Mechanism
In strongly quantum-confined PbS QDs with X-type ligands (halides, thiols, carboxylates), polaron formation occurs due to electrostatic interaction between charge carriers and the negatively charged ligand functional groups. When a charge carrier localizes on a QD, it causes distortion of the Pb-ligand bonds, forming a polaron. Charge transfer between neighboring QDs then requires reorganization of these surface atoms, with associated reorganization energies (λ) ranging from 10s to 100s of meV [14].
When the electronic coupling energy (Vct) between QDs is significantly smaller than this reorganization energy, transport occurs through a phonon-assisted hopping mechanism. The charge transfer rate (kct) in this regime follows the relationship:
$$k{{\mathrm{ct}}} = N{\mathrm{P}}\frac{{2{\uppi}}}{\hbar }V{{\mathrm{ct}}}^2\sqrt {\frac{1}{{4{\uppi}\lambda k{\mathrm{B}}T}}} {\mathrm{e}}^{ - \left( {{\mathrm{\Delta }}E + \lambda } \right)^2/4\lambda k_{\mathrm{B}}T}$$
where NP represents the number of degenerate product states, and ΔE is the energy difference between initial and final states [14]. This hopping mechanism dominates in many QD solids with insulating organic ligands, where carrier mobilities typically remain below 10-2 cm²/Vs.
Band-like Transport in Strongly Coupled QD Solids
With optimized ligand strategies that enhance electronic coupling between QDs, a transition from hopping to band-like transport can occur. When the electronic coupling energy (Vct) becomes sufficiently strong, charge carriers can delocalize across multiple QDs, forming mini-bands that support band-like transport with higher mobilities [71]. The coupling strength depends critically on both the inter-dot distance (controlled by ligand length) and the specific facets exposed on the QD surface. For PbS QDs, calculations show that coupling in the [100] direction is approximately an order of magnitude stronger than in the [111] direction due to carrier confinement away from ligand-rich [111] facets [14].
Table 1: Charge Transport Mechanisms in QD Solids
| Transport Mechanism | Electronic Coupling | Reorganization Energy | Carrier Mobility | Dominant Ligand Types |
|---|---|---|---|---|
| Polaron Hopping | Vct < λ | 10s-100s meV | <10-2 cm²/Vs | Halides, Thiols |
| Band-like Transport | Vct > λ | Minimal contribution | >0.1 cm²/Vs | Conjugated, Inorganic |
Ligand Engineering Strategies for Enhanced Performance
Strategic ligand design has enabled remarkable improvements in QD device performance. The most effective approaches focus on replacing long insulating ligands with shorter or functionally active counterparts.
Short-Chain and Conjugated Organic Ligands
Conjugated ligands with π-electron systems significantly enhance interdot electronic coupling while maintaining passivation. In CsPbI3 perovskite QDs, replacing traditional oleic acid/oleylamine with phenethylammonium iodide (PEAI) using a layer-by-layer solid-state exchange strategy yielded remarkable improvements. This approach enhanced inter-dot coupling, improved defect passivation, and balanced electron and hole transport, resulting in solar cells with a champion PCE of 14.18% with a high open-circuit voltage of 1.23 V [70].
The conjugated phenyl group in PEAI facilitates π-π stacking between adjacent QDs, creating additional pathways for charge transport beyond simple tunneling through insulating barriers. This strategy also improved stability, with unencapsulated devices maintaining performance under high-humidity environments (30-50% relative humidity) due to the hydrophobic nature of the PEA+ cation [70].
Inorganic Ligands and Doping Control
Inorganic ligands such as lead iodide (PbI2) can provide effective passivation while enabling strong interdot coupling. For infrared PbS CQD solar cells, PbI2-passivated QDs exhibit n-type character, which can be strategically manipulated to create bulk heterojunction structures [72]. Reprogramming PbI2-capped CQD surfaces with cysteamine (CTA) ligands converts them to p-type character through a colloid-phase doping strategy, enabling the creation of p-n heterojunctions within the QD solid [72].
This approach allowed the fabrication of thick active layers (exceeding 700 nm) necessary for IR light absorption without compromising open-circuit voltage or fill factor. The resulting devices achieved exceptional charge extraction efficiency exceeding 90% across the ultraviolet to IR range (350-1400 nm) and a significant improvement in Jsc to 37 mA cm-2 under AM1.5 solar illumination [72].
Redox-Active Ligands for Active Charge Transport
A paradigm-shifting approach involves using redox-active ligands that actively participate in charge transport rather than acting as passive spacers. In a groundbreaking demonstration, ZnO QDs were functionalized with ferrocene carboxylate (FcCOO-) ligands, creating electronic states on the QD surface that provide additional pathways for charge transport [4].
These QD/redox ligand assemblies exhibit two complementary charge transport pathways: (1) electron hopping through the QD conduction band and (2) self-exchange through the immobile redox ligands. This dual-path mechanism enables long-range charge transport across the film, with the relative contribution of each pathway tunable through Fermi level positioning and redox ligand coverage [4]. This active ligand strategy represents a significant departure from conventional approaches focused solely on reducing tunneling barriers.
Table 2: Ligand Engineering Strategies and Device Performance
| Ligand Strategy | Material System | Key Findings | Device Performance |
|---|---|---|---|
| PEAI Conjugated Ligand | CsPbI3 PQDs | Enhanced coupling & balanced charge transport | PCE: 14.18%, VOC: 1.23 V |
| PbI2/CTA Doping Control | PbS CQDs (IR) | BHJ structure enables thick active layers | EQE >80% at 1180-1250 nm |
| Ferrocene Redox Ligands | ZnO QDs | Self-exchange chain reaction for long-range transport | Dual-path transport mechanism |
| Zintl-phase BaCd2P2 | Earth-abundant QDs | Defect tolerance, bright photoluminescence (21% QY) | Emerging promising material |
Experimental Protocols for Ligand Exchange and Characterization
Layer-by-Layer Solid-State Ligand Exchange
The layer-by-layer (LBL) deposition method has become the standard for fabricating high-quality QD films with controlled ligand chemistry:
Substrate Preparation: Clean patterned ITO/glass substrates sequentially in detergent, deionized water, acetone, and isopropanol under ultrasonication for 15 minutes each, followed by UV-ozone treatment for 20 minutes [70].
QD Film Deposition: Spin-coat the QD solution (in non-polar solvents like octane) onto the substrate at 2500 rpm for 15 seconds to form a uniform monolayer.
Ligand Exchange: Immediately after deposition, spin-coat the ligand solution (e.g., PEAI in ethyl acetate) at 2000 rpm for 15 seconds to initiate solid-state ligand exchange.
Rinsing: Spin-coat pure solvent (e.g., methyl acetate) at 2000 rpm for 15 seconds to remove excess ligands and reaction byproducts.
Repetition: Repeat steps 2-4 until the desired film thickness (typically 5-10 layers) is achieved [70].
Post-treatment: For some systems, a final post-treatment with ligand solution (e.g., FAI or PEAI in ethyl acetate) is applied, followed by annealing at 70-100°C for 5-10 minutes.
In-Solution Ligand Exchange for Doping Control
Colloid-phase doping enables precise control over carrier type and concentration:
Initial Passivation: Synthesize PbS CQDs with PbI2 passivation to create n-type QDs using established methods [72].
Doping Ligand Introduction: Slowly introduce diluted cysteamine (CTA) solution into stirred PbI2-capped IR CQD inks during continuous stirring.
Doping Optimization: Tune the doping degree by controlling the CTA-to-QD ratio, monitoring the reaction using UV-Vis spectroscopy to maintain size homogeneity.
Purification: Precipitate the doped QDs using non-solvents (e.g., ethyl acetate for PbS CQDs) and centrifuge to obtain pellets.
Redispersion: Redisperse the pellets in appropriate solvents (e.g., octane) to form stable inks for device fabrication [72].
Analytical Characterization Techniques
Comprehensive characterization is essential for linking ligand chemistry to device performance:
Electronic Properties: Cyclic voltammetry determines energy level alignment and charge transfer kinetics in QD/redox ligand assemblies [4].
Structural Analysis: TEM and SEM characterize QD size, morphology, and film homogeneity; XPS quantifies elemental composition and ligand binding [4].
Optical Properties: UV-Vis-NIR spectroscopy monitors absorption features; photoluminescence quantum yield (PLQY) measurements assess defect passivation quality [46].
Charge Transport: Time-resolved microwave conductivity (TRMC) measures carrier mobility; field-effect transistor configurations characterize in-plane transport [46].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for QD Ligand Engineering Research
| Reagent/Category | Function in Research | Example Applications |
|---|---|---|
| Phenethylammonium Iodide | Conjugated short ligand for enhanced coupling & passivation in PQDs | CsPbI3 PQD solar cells & LEDs [70] |
| Lead Iodide (PbI2) | Inorganic passivant providing n-type character to PbS CQDs | IR PbS CQD solar cells [72] |
| Cysteamine | Thiol-based ligand for p-type doping of large-diameter CQDs | p-n Bulk heterojunctions [72] |
| Ferrocene Carboxylate | Redox-active ligand enabling self-exchange charge transport | ZnO QD charge transport studies [4] |
| Formamidinium Iodide | Short ligand for trap passivation in perovskite QDs | Conventional PQD post-treatment [70] |
| Zintl-phase Precursors | Earth-abundant materials for defect-tolerant QDs (Ba, Cd, P) | BaCd2P2 QD synthesis [46] |
Quantitative Performance Relationships
The impact of ligand engineering on device performance can be quantified through several key relationships observed in recent studies:
Ligand Length vs. Mobility: Short-chain ligands reduce interdot distance, exponentially increasing electron hopping rates. For example, reducing ligand chain length from C18 to C8 can improve mobility by 2-3 orders of magnitude [4].
Passivation Quality vs. VOC: Improved surface passivation directly correlates with higher open-circuit voltage in solar cells. The PEAI-treated CsPbI3 PQD devices achieved VOC of 1.23 V, among the highest reported values for this material system [70].
Doping Control vs. JSC: Precise control over carrier density through ligand selection enables optimized built-in fields and charge extraction. The CQD BHJ devices reached JSC of 37 mA cm-2, with significant contributions from the IR region beyond silicon's bandgap [72].
Surface ligand engineering represents the most powerful approach for linking microscopic charge dynamics to macroscopic device performance in quantum dot solids. The strategic design of ligand chemistry—whether through short conjugated molecules, inorganic passivants, or redox-active species—directly controls charge transport mechanisms, enabling unprecedented performance in QD-based optoelectronics.
Future research directions should focus on developing multifunctional ligands that simultaneously address passivation, coupling, and stability requirements. The exploration of Earth-abundant materials like Zintl-phase BaCd2P2 QDs offers promising pathways toward sustainable and scalable QD technologies [46]. Additionally, machine learning-assisted ligand design and advanced in situ characterization will accelerate the discovery of optimal ligand chemistries for specific applications.
As the fundamental understanding of charge transport mechanisms in QD solids continues to mature, ligand engineering will remain the key to unlocking the full potential of these materials for high-performance, solution-processed electronic and optoelectronic devices.
Diagram: Ligand-Mediated Charge Transport Pathways
Ligand Impact on Transport and Performance: This diagram illustrates how different ligand classes dictate charge transport mechanisms in quantum dot solids, ultimately determining device performance metrics such as power conversion efficiency (PCE).
Benchmarking Against Traditional Materials and Setting Performance Targets for Commercialization
Colloidal quantum dots (QDs) represent a significant advancement in semiconductor technology, offering a compelling combination of high performance, solution processability, and customizable optoelectronic properties. For researchers and development professionals, understanding how to benchmark this emerging technology against established traditional materials is crucial for guiding development toward commercial viability. This technical guide provides a comprehensive framework for evaluating quantum dot performance, with particular emphasis on the critical role of surface ligand engineering in modulating charge transport—a fundamental property governing device efficiency and stability.
The core thesis underpinning this analysis posits that active ligand engineering, rather than treating ligands as passive spacers, enables quantum dot solids to not only match but potentially surpass the performance of traditional single-crystal semiconductors in specific applications. This represents a paradigm shift from the traditional view of ligands as mere steric stabilizers toward recognizing them as active components in electronic materials design [4]. Recent research demonstrates that quantum dots can achieve near-unity photoluminescence quantum yields (PLQY) of 99.6%—comparable to the best single-crystal semiconductors—when appropriate measurement techniques and ligand strategies are employed [73].
Performance Benchmarking: Quantum Dots vs. Traditional Semiconductors
Fundamental Material Properties Comparison
Table 1: Performance comparison between quantum dot solids and traditional single-crystal semiconductors
| Performance Parameter | Quantum Dot Solids | Traditional Single Crystals | Measurement Technique | Commercialization Significance |
|---|---|---|---|---|
| Photoluminescence Quantum Yield (PLQY) | Up to 99.6% [73] | >99.9% [73] | Thermal emission analysis [73] | Enables high-efficiency displays and lighting |
| Charge Transport Mechanism | Thermally activated hopping/tunneling [74] | Band transport | Field-dependent conductivity measurements [74] | Determines device architecture and operating conditions |
| Synthetic Requirements | Solution-phase, flask synthesis [73] | Vacuum growth, special conditions [73] | N/A | Lower capital investment, scalable manufacturing |
| Customization Capability | Size-tunable emission wavelengths [73] | Fixed by material composition | Spectroscopy | One material platform for multiple applications |
| Defect Tolerance | Strikingly defect-tolerant [73] | Performance highly defect-sensitive | Comparative emission efficiency | Higher manufacturing yields, reduced purity requirements |
Device-Level Performance Metrics
Table 2: Device performance benchmarks for quantum dot-based optoelectronics
| Device Type | Quantum Dot Performance | Traditional Material Benchmark | Key Ligand Strategy | Commercial Readiness |
|---|---|---|---|---|
| Blue QLEDs | EQE: 24.1%, Luminance: 101,519 cd/m² [75] | Organic LEDs: EQE ~20-30% | Aromatic ligands (3-F-CA) for enhanced interaction [75] | Approaching commercialization with resolution >5000 PPI [75] |
| Quantum Dot Films | Emission: 99.6% of absorbed light [73] | Single crystals: >99.9% | Eight atomic layer coating for superior quality [73] | Ready for display enhancement films |
| QD/Redox Ligand Assemblies | Two complementary charge transport pathways [4] | Single transport mechanism | Ferrocene carboxylate ligands enabling self-exchange [4] | Research stage with potential for specialized sensors |
The Central Role of Surface Ligands in Charge Transport Engineering
Ligand Length and Electronic Coupling
The length of surface ligands directly governs interparticle distance in quantum dot solids, exponentially affecting the electron tunneling rate between adjacent dots. Short-chain ligands like 3-fluorocinnamate (3-F-CA) dramatically enhance carrier mobility and conductivity compared to long-chain aliphatic ligands such as oleic acid (OA) [75]. This enhancement is quantitatively demonstrated in device performance, where QLEDs with 3-F-CA ligands achieve external quantum efficiencies of 24.1%—more than double the efficiency of devices with OA ligands (7.7%) [75]. Beyond merely reducing physical separation, shorter ligands decrease the tunneling barrier width, enabling more efficient charge carrier injection and extraction in operational devices.
The relationship between ligand structure and electronic properties can be visualized through the following charge transport mechanisms:
Beyond Passive Spacers: Active Redox Ligands
A revolutionary approach in ligand engineering involves shifting from passive spacer ligands to actively functioning redox ligands that introduce electronic states and provide dedicated paths for carrier transfer [4]. This strategy employs ligands with well-defined redox states that can participate directly in charge transport processes through self-exchange chain reactions, enabling long-range charge transport across quantum dot films.
In engineered systems using ferrocene carboxylate (FcCOO-) ligands on ZnO quantum dots, charge transport occurs through two complementary pathways: (1) electron hopping through the conduction band of the QDs, and (2) self-exchange through the immobile redox ligands [4]. This dual-pathway mechanism demonstrates that appropriately designed ligand systems can actively participate in rather than merely facilitate charge transport. The kinetics of these processes can be systematically controlled through modulation of the Fermi level and redox ligand coverage, following predictions of percolation theory [4].
Experimental Protocols for Charge Transport Characterization
Field-Dependent Conductivity Measurements
Understanding charge transport in quantum dot solids requires specialized characterization techniques that account for their unique electronic properties. The following workflow outlines standard protocols for evaluating field-dependent conductivity:
Detailed Methodology:
- Device Fabrication: Prepare quantum dot solid films with controlled thickness (typically 100-500 nm) using spin-coating or drop-casting methods. Implement ligand exchange procedures by immersing films in solutions containing target ligands (e.g., 0.7 μmol 3-F-CA in toluene for 12 hours) followed by thorough rinsing to remove unbound ligands [75].
- Electric Field Application: Apply electric fields across the film in the range of 1-10 V/μm, which represents typical operational conditions for quantum dot photovoltaic and light-emitting devices [74].
- Temperature-Dependent Measurements: Conduct measurements at varying temperatures (typically 10-300 K) to distinguish between different charge transport mechanisms.
- Data Interpretation: Analyze the resulting current-voltage characteristics using heat balance models that describe the increased effective electronic temperature (T_eff) that exceeds that of the lattice under applied fields. Fit data to established models to extract key parameters such as localization length (typically 2-5 nm for ZnO QDs) and activation energies [74].
Electrochemical Characterization of Redox Ligand Systems
For quantum dot solids incorporating redox-active ligands, electrochemical techniques provide unique insights into charge transport mechanisms:
Cyclic Voltammetry Protocol:
- Assembly Preparation: Develop experimental models consisting of quantum dots with anchored redox ligands (e.g., ZnO QDs with ferrocene carboxylate ligands). Ensure even distribution of redox ligands across the film, confirmed through XPS depth profiling [4].
- Three-Electrode Configuration: Immerse QD/redox ligand assemblies in electrolyte solution (e.g., acetonitrile with tetrabutylammonium hexafluorophosphate) using standard three-electrode electrochemical cell.
- Fermi Level Scanning: Scan the Fermi level while recording current due to injected/extracted charges. Typical parameters: scan from -0.6 V to -1.7 V (vs Fc/Fc+), revert to +0.9 V, then return to initial value at scan rates of 10-100 mV/s [4].
- Signal Deconvolution: Identify characteristic signals in the voltammogram corresponding to (i) charge injection into the conduction band, (ii) quasi-reversible redox ligand activity, and (iii) additional charge transfer processes.
The Researcher's Toolkit: Essential Materials and Reagents
Table 3: Essential research reagents for quantum dot ligand studies
| Reagent Category | Specific Examples | Function | Experimental Considerations |
|---|---|---|---|
| Short-Chain Aromatic Ligands | 3-fluorocinnamate (3-F-CA) | Enhance interdot interaction via π-π stacking, improve charge transport | Optimal binding at 0.7 μmol amount; increases PLQY to 93% [75] |
| Redox-Active Ligands | Ferrocene carboxylate (FcCOO-) | Introduce active charge transport pathways via self-exchange reactions | Achieve ~10 ligands per QD; even distribution confirmed by XPS [4] |
| Aliphatic Ligands | Oleic Acid (OA), 3-mercaptopropionic acid (MPA) | Reference spacers; modulate tunneling distance | MPA-CdTe QDs show stronger mitochondrial effects than TGA-CdTe [76] |
| QD Core Materials | CdZnSe, ZnO, CdTe | Photovoltaic, sensing, or display applications | ZnO QDs with FcCOO- enable electrochemical studies [4] |
| Characterization Tools | Atomic layer deposition (ALD) SnO₂ | Buffer layers for efficiency and stability enhancement | Increases shunt resistance in perovskite solar cells [77] |
Performance Targets for Commercialization
Efficiency and Stability Milestones
Based on current benchmarking data, the following performance targets represent thresholds for commercial viability in key application areas:
Display Applications: External quantum efficiency (EQE) >20% for blue QLEDs with luminance >100,000 cd/m² and operational stability T95 >1000 hours at 1000 cd/m² [75]. Current state-of-the-art blue QLEDs achieve EQE of 24.1% with extrapolated T95 lifetime of 54 hours at 1000 cd/m² [75].
Photovoltaic Applications: Photoluminescence quantum yield (PLQY) >99.9% with field-dependent conductivity maintaining optimal performance at operational fields of 1-10 V/μm [73] [74]. Current research demonstrates PLQY of 99.6% with potential for further improvement through advanced ligand engineering.
General Optoelectronics: Defect-tolerant charge transport with localization lengths of 2-5 nm, enabling efficient conduction through quantum dot solids with minimal performance degradation due to structural imperfections [74].
Manufacturing and Scalability Considerations
Commercialization requires not only performance benchmarks but also manufacturing feasibility:
Ligand Exchange Processes: Development of scalable ligand exchange techniques that ensure complete surface functionalization with high reproducibility. Current methods include solution-phase exchange and solid-state ligand exchange, each with distinct advantages for specific applications.
Patterned Array Fabrication: Achievement of long-range ordered quantum dot arrays with minimum pixel size ≤3 μm enabling resolution >5000 PPI for display applications [75]. Aromatic-enhanced capillary bridge confinement strategies have demonstrated feasibility of this target.
Stability Standards: Implementation of encapsulation strategies and buffer layers (such as ALD-processed SnO₂) that enable devices to maintain >90% of initial efficiency after 600 hours storage in ambient conditions (20-40% relative humidity) without encapsulation [77].
The benchmarking data presented demonstrates that quantum dot solids, when appropriately engineered with optimized surface ligands, can indeed compete with traditional single-crystal semiconductors across multiple performance parameters. The strategic application of short-chain conjugated ligands and redox-active ligands has enabled remarkable progress in closing the performance gap while maintaining the processing advantages of solution-phase materials.
Future research directions should focus on further elucidating the structure-property relationships governing charge transport in heterogeneous quantum dot solids, developing increasingly sophisticated ligand systems that provide multiple functionalities (including self-healing properties and environmental stability), and refining manufacturing processes to enable uniform ligand distribution in large-area devices. The continued refinement of performance targets based on application-specific requirements will ensure that research efforts remain aligned with commercial needs, accelerating the adoption of quantum dot technologies across diverse electronic and optoelectronic applications.
Conclusion
The strategic engineering of surface ligands has evolved from a simple process of shortening molecular chains to a sophisticated discipline of designing active molecular components that directly participate in charge transport. The key takeaway is that optimal performance in quantum dot solids is achieved not by minimizing ligand presence, but by judiciously selecting ligands that provide effective surface passivation while simultaneously enabling efficient charge delocalization through enhanced electronic coupling or novel mechanisms like redox-mediated self-exchange. Future progress hinges on the development of multifunctional ligands that combine excellent charge transport with enhanced stability and specific targeting capabilities. For biomedical research, this opens avenues for designing highly sensitive QD-based biosensors that operate on charge transfer principles and for creating novel theranostic platforms where charge separation efficiency directly impacts imaging fidelity or therapeutic activation. The continued synergy between advanced synthesis, precise spectroscopic validation, and device engineering will be paramount in translating these material breakthroughs into clinical and commercial realities.
References
- 1. Charge transport in functional ligand capped nanocrystals ... [https://www.sciencedirect.com/science/article/abs/pii/S2211285525007104]
- 2. Ultrafast charge transfer dynamics in lead sulfide quantum ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra00479a]
- 3. The role of surface ligands in determining the electronic ... [https://pubs.rsc.org/en/content/articlelanding/2018/nr/c7nr06116a]
- 4. Long-Range Charge Transport via Redox Ligands in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9798906/]
- 5. Enhanced charge carrier transport properties in colloidal ... [https://pubs.rsc.org/en/content/articlehtml/2016/ta/c6ta06835a]
- 6. Engineering the Surface Chemistry of Colloidal InP Quantum ... [https://imod-stc.org/publication/engineering-the-surface-chemistry-of-colloidal-inp-quantum-dots-for-charge-transport/]
- 7. Spatially resolved charge-transfer kinetics at the quantum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11962476/]
- 8. 《Joule》刊发材料科学与工程学院张晓亮教授课题组最新研究 ... [http://www.mse.buaa.edu.cn/info/1019/5845.htm]
- 9. Preparation of sulfur group compounds and perovskite ... [https://www.sciencedirect.com/science/article/pii/S2589965125000248]
- 10. Quantum dot-based non-volatile memory - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d4ra08307e]
- 11. Impact of Different Surface Ligands on the Optical ... [https://www.mdpi.com/1996-1944/8/4/1858]
- 12. Engineering colloidal quantum dot solids within and ... [https://www.nature.com/articles/ncomms4803]
- 13. Colloidal quantum dot for infrared-absorbing solar cells [https://www.sciopen.com/article/10.26599/NRE.2023.9120095]
- 14. Charge transport in semiconductors assembled from ... [https://www.nature.com/articles/s41467-020-16560-7]
- 15. Charge Injection and Energy Transfer of Surface ... [https://www.mdpi.com/2079-4991/13/7/1159]
- 16. Charge dynamics of individual conductance channels ... [https://arxiv.org/html/2510.01162v1]
- 17. Elementary Charge Characterization of Single Quantum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412147/]
- 18. HSAB theory [https://en.wikipedia.org/wiki/HSAB_theory]
- 19. 3.2.1: Hard and Soft Acid and Base Theory [https://chem.libretexts.org/Courses/Saint_Marys_College_Notre_Dame_IN/CHEM_342%3A_Bio-inorganic_Chemistry/Readings/Week_3%3A_Metal-Ligand_Interactions_continued..../3.2%3A_The_identity_of_metal_ion_and_the_ligand_donor_atom(s)_affects_affinity/3.2.1%3A_Hard_and_Soft_Acid_and_Base_Theory]
- 20. Development of the design and synthesis of metal–organic ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d4cs00432a]
- 21. The effects of PbS quantum dot surface contributing to their ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894724085723]
- 22. Quantum Dot-Based Electron-Transporting Materials for ... [https://pubmed.ncbi.nlm.nih.gov/40988598/]
- 23. Rational ligand design for enhanced carrier mobility in self ... [https://pubs.rsc.org/en/content/articlehtml/2024/nh/d4nh00038b]
- 24. Quantum dots coordinated with conjugated organic ligands [https://pmc.ncbi.nlm.nih.gov/articles/PMC3246356/]
- 25. AFM-IR of Electrohydrodynamically Printed PbS Quantum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11378332/]
- 26. Morphological consequences of ligand exchange in ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119917306146]
- 27. Observation of ordered organic capping ligands on ... [https://www.nature.com/articles/s41467-021-22947-x]
- 28. An 'impossible' 20-electron molecule challenges 100 years ... [https://www.sciencedaily.com/releases/2025/07/250727235814.htm]
- 29. Impact of Different Surface Ligands on the Optical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5507044/]
- 30. Hybrid passivated colloidal quantum dot solids [https://pubmed.ncbi.nlm.nih.gov/22842552/]
- 31. Enhanced charge carrier transport properties in colloidal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5735354/]
- 32. Research progress on surface ligand engineering and size ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775325005981]
- 33. Improving the charge transport of perovskite nanocrystal ... [https://www.sciencedirect.com/science/article/pii/S2666523925000078]
- 34. Lead(II) iodide [https://en.wikipedia.org/wiki/Lead(II)_iodide]
- 35. Lead Iodide (PbI2) Semiconductors [https://www.azom.com/article.aspx?ArticleID=8418]
- 36. Recent Research Progress in Surface Ligand Exchange of ... [https://www.mdpi.com/2076-3417/10/3/975]
- 37. THE CRYSTAL STRUCTURE OF LEAD(II) IODIDE- ... [https://www.semanticscholar.org/paper/THE-CRYSTAL-STRUCTURE-OF-LEAD%28II%29-2%29%2C-PbI2%28dmso%292-Miyamae-Numahata/b0759943bb5fa045b375d9e6ecbb465873cfb056]
- 38. Improved performance and stability in quantum dot solar ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4110173/]
- 39. Ligand-customized colloidal quantum dots for high- ... [https://pubs.rsc.org/en/content/articlelanding/2024/tc/d4tc01182a]
- 40. Ligand-induced charge transport modulation and ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433223002994]
- 41. Charge dynamics of individual conductance channels ... [https://arxiv.org/abs/2510.01162]
- 42. Non-Radiative Carrier Recombination Enhanced by Two- ... [https://www.nature.com/articles/srep21712]
- 43. [PDF] Impact of Different Surface on the... | Semantic Scholar Ligands [https://www.semanticscholar.org/paper/Impact-of-Different-Surface-Ligands-on-the-Optical-Xu-Gerlein/e978e02925367037209f35cac6734dab1ac7d6d6]
- 44. Injection and Energy Transfer of Charge -Engineered... Surface [https://pmc.ncbi.nlm.nih.gov/articles/PMC10096696/]
- 45. Photoluminescence characterization of non-radiative ... [https://www.sciencedirect.com/science/article/abs/pii/S0040609099009554]
- 46. Built for Brilliance: Zintl-Phase Quantum Dots Illuminate ... [https://www.nrel.gov/news/detail/program/2025/built-for-brilliance-zintl-phase-quantum-dots-illuminate-new-opportunities-for-optoelectronics]
- 47. Counterion docking: a general approach to reducing ... [https://www.nature.com/articles/s41467-024-49208-x]
- 48. Realizing low voltage-driven bright and stable quantum dot ... [https://www.nature.com/articles/s41377-024-01727-4]
- 49. Probing Dopant Size Effects on Defect Clustering and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412159/]
- 50. PbTe quantum dots highly packed monolayer fabrication by a ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0317677]
- 51. The relationship between the length of surface ligand and ... [https://www.sciencedirect.com/science/article/abs/pii/S0045653517309943]
- 52. Automated Quantum Dots Purification via Solid Phase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9230973/]
- 53. Low-temperature molten-salt enabled synthesis of highly ... [https://www.nature.com/articles/s41467-025-63653-2]
- 54. Air exposure oxidation and photooxidation of solution- ... [https://www.sciencedirect.com/science/article/abs/pii/S0927024819304921]
- 55. Ligand dependent oxidation dictates the performance ... [https://pubs.rsc.org/en/content/articlehtml/2020/se/c9se00602h]
- 56. Single-step, conformal, and efficient assembly of ligand ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr04620j]
- 57. Double Metal Oxide Electron Transport Layers for Colloidal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7221608/]
- 58. Understanding of Charge Transport and Non-Linear ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119925000850]
- 59. Reshaping quantum dots production through continuous ... [https://www.sciencedaily.com/releases/2025/04/250411175713.htm]
- 60. Machine learning models for accurately predicting ... [https://www.nature.com/articles/s41598-025-08110-2]
- 61. Attosecond formation of charge-transfer-to-solvent states ... [https://www.nature.com/articles/s41467-024-52740-5]
- 62. Ultra-Fast Charge Transfer in P3HT Composites Using the ... [https://www.mdpi.com/2079-4991/15/6/433]
- 63. Attosecond formation of charge-transfer-to-solvent states ... [https://pubmed.ncbi.nlm.nih.gov/39406706/]
- 64. The effect of surface ligands on the surface chemical states ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra05099e?page=search]
- 65. Colloidal Quantum Dot Inks for Single-Step-Fabricated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5814956/]
- 66. Research Review Quantum dot field effect transistors [https://www.sciencedirect.com/science/article/pii/S1369702113002897]
- 67. Enhancing charge extraction efficiency in PbS-I quantum ... [https://www.sciencedirect.com/science/article/abs/pii/S0038092X2500012X]
- 68. In situ 2D-perovskite-like ligands offer versatile passivation of ... [https://pubs.rsc.org/en/content/articlehtml/2025/el/d5el00135h]
- 69. Solid‐State Ligand‐Exchange Fabrication of CH3NH3PbI3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5067684/]
- 70. Modified surface ligand management of CsPbI 3 perovskite ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119924000570]
- 71. Charge transport in strongly coupled quantum dot solids [https://pubmed.ncbi.nlm.nih.gov/26551016/]
- 72. Colloidal Quantum Dot Bulk Heterojunction Solids with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7404161/]
- 73. Quantum Dots Vs. Crystal Semiconductors: The Future ... [https://www.aau.edu/research-scholarship/featured-research-topics/quantum-dots-vs-crystal-semiconductors-future-scifi]
- 74. Strongly Electric Field Dependent Conductivity in Quantum ... [https://arxiv.org/abs/2511.14212]
- 75. Long-range order enhance performance of patterned blue ... [https://www.nature.com/articles/s41467-025-62345-1]
- 76. The relationship between the length of surface ligand and ... [https://pubmed.ncbi.nlm.nih.gov/28672691/]
- 77. Tin dioxide buffer layer-assisted efficiency and stability of wide ... [https://www.jos.ac.cn/article/id/a7797872-29a0-46f1-a0a7-9ec324a59081]